未来早知道:用Keras的深度学习预测疾病
点击蓝字
关注小图
深度学习就像是机器学习的炫酷版本!它是一种让机器变得超级聪明的黑科技。通过模拟人脑神经网络的方式,深度学习模型可以从大量数据中学习并做出准确的预测。就像小宝宝学习走路一样,深度学习模型也是通过不断练习和试错来提升自己的能力。它可以识别图像、翻译语言、自动驾驶,甚至可以为我们写诗!总之,深度学习让机器变得像人一样聪明,带来了无数有趣和实用的应用。
Keras(https://keras.io/)就像是深度学习的瑞士军刀!它是一个酷炫且好用的工具,可以帮助你构建和训练神经网络。不管你是一个新手还是一个专业人士,Keras都会给你带来惊喜!想象一下,你想要建立一个神经网络模型,但是不想从头开始写一大堆繁琐的代码。这时候,Keras就派上了用场!它提供了一种简单而强大的方式来创建神经网络。你只需要一些简单的代码,就可以像搭积木一样组装你的模型。
不仅如此,Keras还有一堆酷炫的功能。它支持各种常见的神经网络层,比如全连接层、卷积层、循环层等。你只需要选择合适的层,然后将它们堆叠在一起,就能构建出你想要的模型。而且,Keras还支持多种优化器和损失函数,这些是训练模型时必不可少的东西。你可以根据你的任务和喜好来选择合适的优化器,让你的模型训练得更快、更准确。
最酷的是,Keras不仅易于使用,而且还有一个庞大的社区支持。你可以在社区里找到各种各样的例子和帮助。还有很多有趣的教程和博客,可以让你在深度学习的世界里畅游。
总而言之,Keras就是一个酷炫实用的深度学习工具,帮助你构建强大的神经网络。不管你是深度学习新手还是高手,Keras都是你的好朋友,让你轻松愉快地探索深度学习的奇妙世界!
Keras主页

以下是一个简单的示例,展示如何使用R包keras进行糖尿病预测模型的训练和评估:
代码具体包括:
Step1 导入数据集以及数据预处理
# 这里的"inputdata.txt"是自行准备的本地文件,小图给大家附在最后。# 1.导入数据集以及数据预处理# 安装和加载所需的库library(keras)# 导入数据集data <- read.table(file = "D:/wanglab/life/ziyuan/20230628/inputdata.txt",header = T) # 替换为实际数据集文件的路径# 数据预处理# 数据标准化data[, 1:8] <- scale(data[, 1:8])# 将目标变量转换为因子data$diabetes <- as.factor(data$diabetes)# 划分数据集为训练集和测试集set.seed(123) # 设置随机种子,保证结果可复现indices <- sample(1:nrow(data), nrow(data)*0.8) # 80%的数据用于训练,20%的数据用于测试train_data <- data[indices, ]test_data <- data[-indices, ]
Step2创建深度学习模型
# 2.创建深度学习模型model <- keras_model_sequential() # 创建序贯模型model %>%layer_dense(units = 32, activation = "relu", input_shape = 8) %>% # 添加一个全连接层,包含32个神经元,使用ReLU激活函数layer_dense(units = 16, activation = "relu") %>% # 添加第二个全连接层,包含16个神经元,使用ReLU激活函数layer_dense(units = 1, activation = "sigmoid") # 添加输出层,使用Sigmoid激活函数,输出为二进制分类结果
Step3编译模型
# 3.编译模型model %>% compile(loss = "binary_crossentropy", # 使用二元交叉熵作为损失函数optimizer = "adam", # 使用Adam优化器进行参数优化metrics = c("accuracy") # 指定评估指标为准确率)
Step4训练模型
# 4.训练模型history <- model %>% fit(x = as.matrix(train_data[, 1:8]), # 输入特征y = as.numeric(train_data$diabetes) - 1, # 目标变量(将因子转换为数值)epochs = 10, # 迭代次数batch_size = 32, # 批处理大小validation_split = 0.2 # 将训练集的20%用于验证)
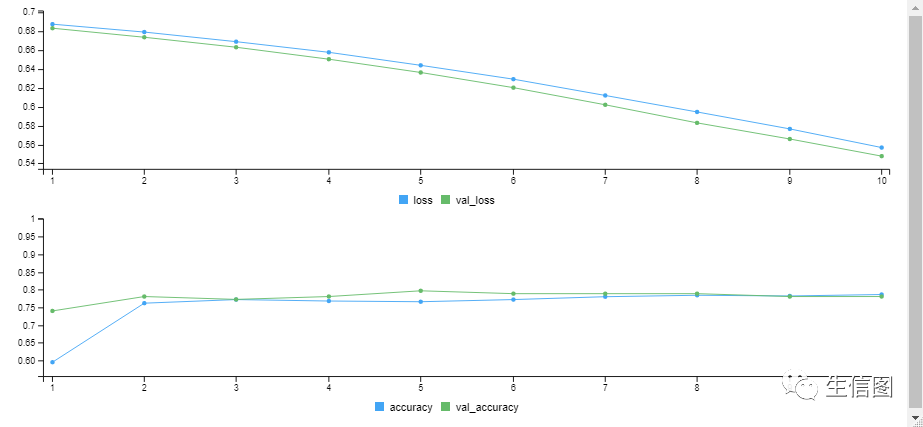
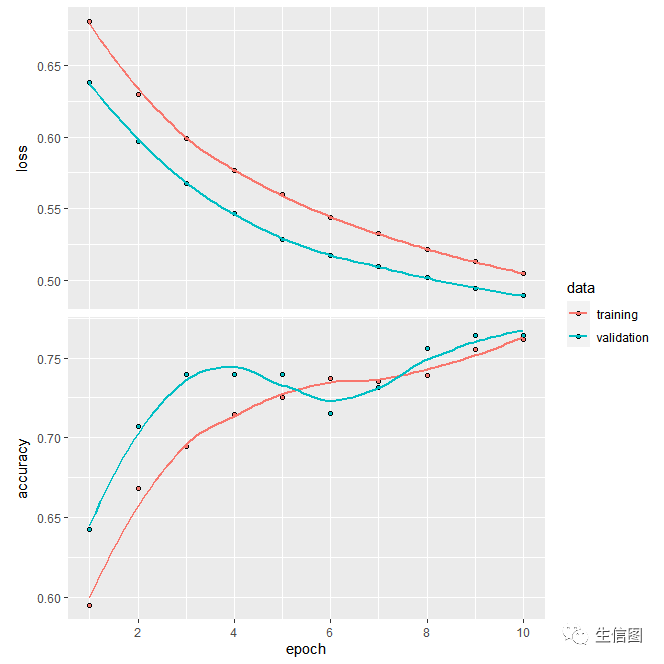
Step5评估模型
# 5.评估模型eval <- model %>% evaluate(x = as.matrix(test_data[, 1:8]), # 测试集的输入特征y = as.numeric(test_data$diabetes) - 1, # 测试集的目标变量(将因子转换为数值)verbose = 0 # 不显示详细信息)cat("Test Accuracy:", eval, "n")eval[2]# 绘制训练过程中的准确率和损失变化曲线plot(history)
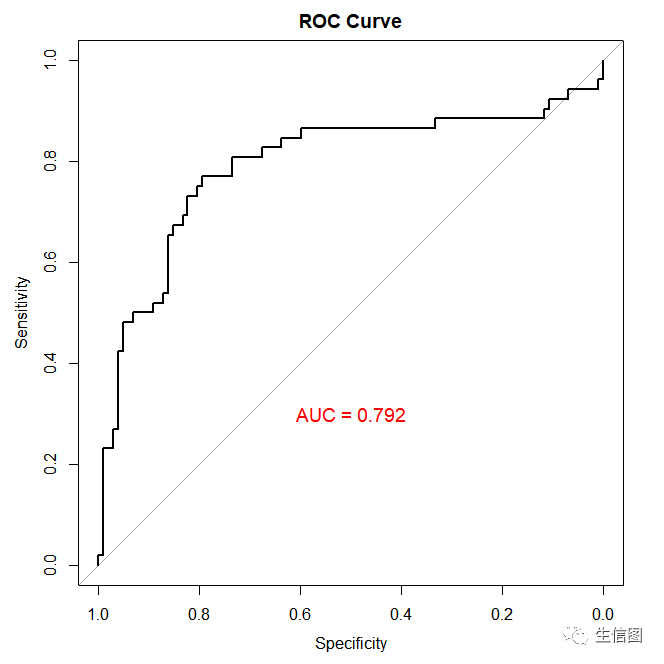
Step6绘制AUC曲线
# 预测概率predictions <- model %>% predict(as.matrix(test_data[, 1:8]))predicted_classes <- ifelse(predictions > 0.5, 1, 0) # 将概率转换为类别预测(大于0.5为正类,否则为负类)# 计算AUClibrary(pROC)roc_obj <- roc(test_data$diabetes, predictions)auc_value <- auc(roc_obj)# 绘制AUC曲线plot(roc_obj, main = "ROC Curve")text(0.5, 0.3, paste0("AUC = ", round(auc_value, 3)), col = "red", cex = 1.2)
注:
# 请确保将路径”inputdata.txt”替换为实际数据集文件的路径,并根据需要调整模型参数和其他配置。
# 在绘制AUC曲线之前,确保您的模型输出的是预测的概率值,而不是类别标签。
厉害吧,深度学习在R语言中的实现非常简单。我们使用的都是入门级函数,只需对R语言有一定了解,就可以轻松实现啦。
需要注意的是,如果要使用深度学习进行分析,并且特征值较多,可能需要大量的内存。在本地电脑上会面临内存不足的问题。因此,建议使用服务器进行分析。然而,租用服务器的费用可能较高。为了解决这个问题,我们建议使用我们的云生信平台,该平台提供强大的计算资源和深度学习工具。您可以访问我们的云生信平台,链接为:http://www.biocloudservice.com/home.html。
欢迎使用:云生信平台 ( http://www.biocloudservice.com/home.html)

|
往期推荐 |
|
|
|
|
|
|
👇点击阅读原文进入网址