mlr3中的机器学习–进入生物信息学新时代(1)
点击蓝字 关注我们
今日话题
mlr3中的机器学习–进入生物信息学新时代
NEWS
”
机器学习在生物信息学领域的发展具有重要意义,它已为生物学研究和医学领域带来了深远的影响。
高通量技术的发展使得生物学家可以生成大量的生物数据,包括基因组、蛋白质组、转录组等信息。机器学习算法能够有效处理这些海量数据,从中挖掘隐藏的模式和关联,帮助研究人员更好地理解生物体系。该方法能够根据已知的生物数据,建立模型来预测未知的结果。
在生物学数据中,往往包含大量的特征(如基因或蛋白质)。机器学习可以帮助生物学家选择最相关的特征,从而降低维度并提高模型的性能,减少过拟合的风险。
目前,在生物信息学中,机器学习有了新的应用和拓展。例如,可以利用已知的基因表达数据,预测患者是否患有某种疾病,或者预测蛋白质的结构和功能。这对于药物研发、疾病诊断和治疗等方面具有
NEWS
”
重要意义:
1 药物发现:
通过分析已知的药物与基因、蛋白质相互作用的数据,机器学习可以预测新的药物与生物分子之间的相互作用,从而加速药物发现过程。
2 个性化医疗:
机器学习在个体基因组和临床数据分析中的应用,使医疗可以更加个性化和精准。例如,根据患者的基因信息,可以预测患者对特定药物的反应,从而指导医疗决策。
3 生物图像分析:
生物图像数据,如显微镜图像和医学影像,可以通过机器学习算法进行分析和解释。这可以用于细胞结构识别、肿瘤检测等应用。
4 基因组学研究:
机器学习在基因组学中可以用于预测基因调控、DNA序列分析、基因表达模式识别等任务,有助于揭示基因与表型之间的关联。
NEWS
”
总而言之,机器学习的发展为生物信息学领域带来了工具和方法,能够加速科学研究的进程,推动医学进步,并为个性化医疗和精准治疗提供了新的可能性。它已经成为生物学家和医学研究人员不可或缺的工具之一。
NEWS
”

mlr3 是一个在R语言中实现的强大机器学习框架,专注于使机器学习的工作流程更加高效、灵活和可扩展。它是 mlr 包的继任者,是一个全面重新设计和重构的版本,旨在提供更好的性能和易用性。mlr3 提供了丰富的功能,用于数据预处理、模型选择、调参和评估等机器学习任务。今天,小果就带大家走进先进的mlr3R包,首先从mlr3中的task对象说起,在 mlr3 中,Task 是一个重要的对象,用于定义和管理机器学习问题的输入数据和输出目标。每个 Task 对象表示一个机器学习任务,例如分类、回归或生存分析等。Task 对象存储了特征矩阵和对应的目标向量,以及一些元数据,为模型训练和评估提供了基础。
首先,我们需要安装mlr3包并导入。
NEWS
”
install.packages("mlr3")library(mlr3)
mlr3包在mlr_tasks字典中储存了预定义的机器学习任务,如波士顿房价的预测,mtcars,iris花的分别与识别等非常常见的机器学习任务:
mlr_tasks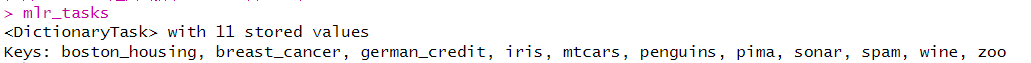
可以使用tsk()函数从该mlr_tasks中获取任务,并将返回值分配给新的变量。
tsk_mtcars = tsk("mtcars")tsk_mtcars

一.构造task
有时,我们想要创建属于自己的回归任务,怎么办呢?可以构造一个新的TaskRegr实例,在下面的例子中,小果向大家给出了一个构建TaskRegr实例的例子,首先加载数据集,将数据子集化为仅包含三个列(对应三个需要的变量)并打印修改后数据的属性,然后再设置一个名为tsk_mtcars的回归任务,在该任务中,小果将带大家尝试着提供过气缸数和位移量去预测汽车的每加仑英里。
在这个问题中,主要的函数时as_task_regr()函数,as_task_regr() 函数允许你将数据集和连续数值目标转化为适用于回归建模的 Task 对象,这样就可以使用 mlr3 中的回归学习算法对数据进行建模和预测。
data("mtcars", package = "datasets")mtcars_subset = subset(mtcars, select = c("mpg", "cyl", "disp"))str(mtcars_subset)
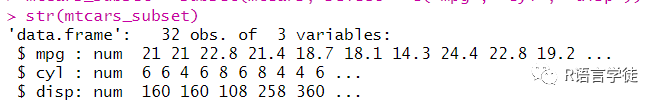
首先,使用 data() 函数加载了 R 中自带的 datasets 包中的 mtcars 数据集,该数据集包含了关于汽车的一些特征和性能指标。然后,代码使用 subset() 函数从 mtcars 数据集中选择了一些列作为子集。这个子集只包含了三个列,即 “mpg”(每加仑英里数)、”cyl”(汽缸数)和 “disp”(排量)。str() 函数被用来查看 mtcars_subset 数据框的结构,这是为了观察数据的列和属性。我们可以看到,mtcars_subset是一个具有三个特征变量的截面数据。
tsk_mtcars = as_task_regr(mtcars_subset, target = "mpg", id = "cars")#创建 在这段代码中,我们创建了回归任务的 Task 对象。使用 as_task_regr() 函数,将之前创建的 mtcars_subset 数据框转换为回归任务的 Task 对象。参数包括:
data:输入数据框,这里是 mtcars_subset。
target:指定了目标变量,也就是我们要预测的变量,这里是 “mpg”。
id:可选的任务标识符,这里设置为 “cars”。
这样,通过该代码,我们就创建了一个名为 tsk_mtcars 的回归任务的 Task 对象,其中特征矩阵是来自于 mtcars_subset 数据框的 “cyl” 和 “disp” 列,目标变量是 “mpg” 列,任务标识符设置为 “cars”。
tsk_mtcars#输出任务摘要 在创建完任务后,可以通过在命令行中输入任务名来获取任务摘要:

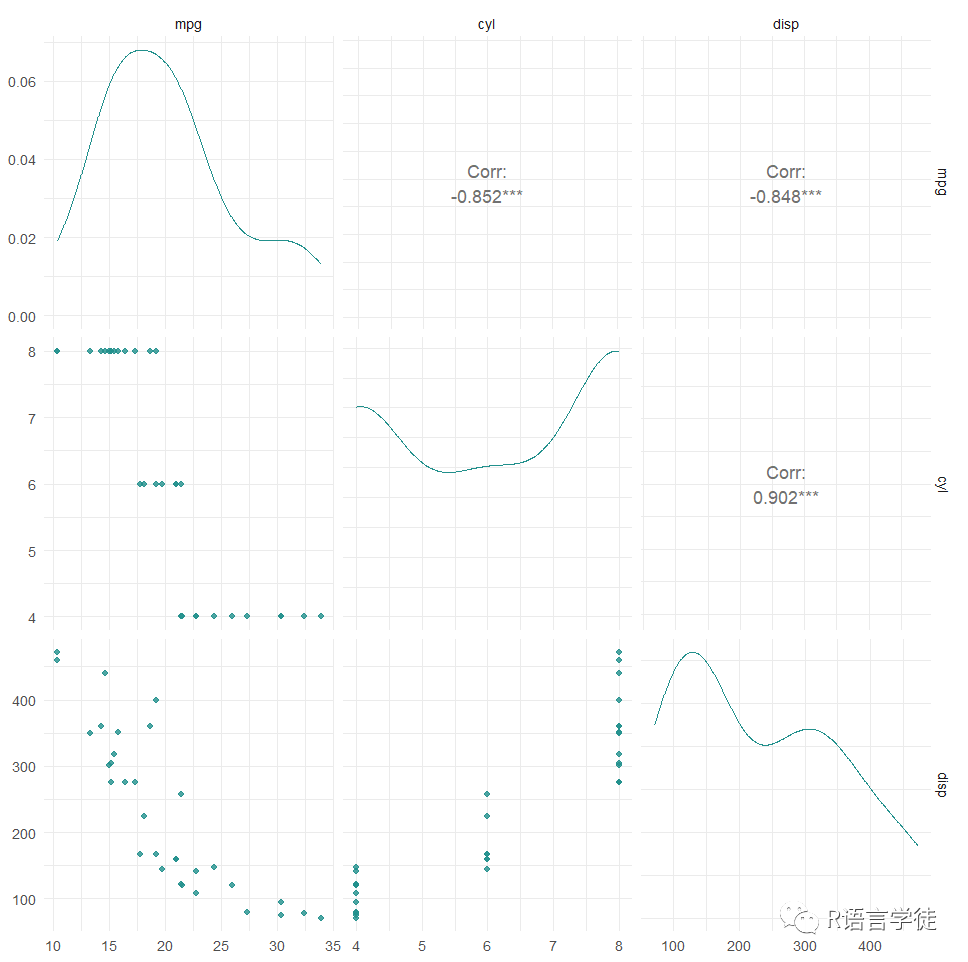
也可以使用mlr3viz包对任务进行绘制,给出目标变量和特征变量分布的图形摘要,在这张图中,我们可以看到任务中三个变量的具体分布:
library(mlr3viz)autoplot(tsk_mtcars, type = "pairs")#可视化任务
二.检索数据
小师妹已经教会了大家如何创建任务来存储数据,那么下面,小师妹将带领大家检索存储的数据。
可以使用$nrow、$ncol来检索数据的维度,分别为任务中数据框的行数(通常代表样本容量)和列数(通常代表维度的个数):
c(tsk_mtcars$nrow, tsk_mtcars$ncol)#检索数据维度 
可以通过$feature_names和$target_names来获取特征变量和响应变量的具体名称:
c(Features = tsk_mtcars$feature_names,Target = tsk_mtcars$target_names)#获取特征和响应变量的变量名

可以通过$row_ids来获取行ID,行ID可以访问单个观测值的数据,与行号不同,行ID由自然数表示,它是任务行的名称,代表样本的标号:
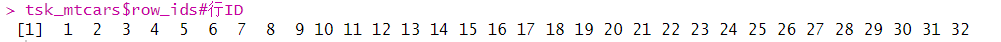
tsk_mtcars$row_ids#行IDtask=tsk_mtcarstask$filter(c(4, 1, 3))task$row_ids
上面的代码表示通过行ID去选取任务中对应的样本:
task$filter(c(4, 1, 3)):使用 filter() 方法来对任务对象进行筛选操作。参数 c(4, 1, 3) 是一个索引向量,它指定了要保留的样本的索引。在这个例子中,它表示保留数据中索引为 4、1 和 3 的样本。筛选后的结果会反映在任务对象 task 中。
task$row_ids:这行代码获取任务对象中所有样本的行索引(行标识符)。
代码通过对回归任务的数据进行筛选,保留了索引即样本标号为 4、1 和 3 的样本。可以通过查看 task$row_ids 来获取筛选后的样本的行索引列表。这个操作在一些情况下可以帮助对数据集进行子集的操作,以便于特定的分析和建模需求。(这种设计决策允许任务和学习者在真正的数据库管理系统上透明地操作,其中主键要求是唯一的,但不一定是连续的。)
在task中,相应的数据可以通过$data()进行访问将返回一个data.table对象,该方法可以选择相应的参数,用来指定要检索的数据子集:
tsk_mtcars$data()#检索所有数据tsk_mtcars$data(rows = c(1, 5, 10), cols = tsk_mtcars$feature_names)#检索需要的数据

到这里,task对象的基本属性和有关操作小师妹就向大家讲完了,不知道同学们是否学会了呢,在小师妹的陪伴下,相信小伙伴们已经基本掌握了mlr3中task任务的相关知识,会通过R语言编写相应的程序,小师妹要和大家说再见咯,一定要自己练习一下哦,同时如果大家想要继续了解更多有关R语言内容可以持续关注小师妹哦~~或者也可以关注我们的官网也会持续更新的哦~ http://www.biocloudservice.com/home.html

