小果三分钟教会你如何对Trinity转录本进行下游分析(下)
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

在上一期的教学中小果讲到了如何下载和使用Trinity组装转录本,并且评估转录本的质量,在本期中我们将当组装完成后可以进行那些下游分析来挖掘我们关注的生物学问题。
跟着小果一起来看看吧
1.转录本丰度
在进行其它分析之前,我们通常学对转录本和基因的丰度进行定量。在Trinity中提供了调用三种不同软件的脚本,RSEM,kallisto和salmon。值得注意的是Trinity并没有同时打包安装好这三个软件,需要我们自己下载并配置好相对应的环境变量。
我们使用一下面的脚本perl脚本进行计算
$ TRINITY_HOME/util/align_and_estimate_abundance.pl主要参数如下:
# --transcripts <string>: 转录本文件# --seqType <string>: fq|fa# 若为paired-end数据:--left <string>: 左端reads文件 --right <string>: 右端reads文件# single-end数据:--single <string>: 原始reads文件# --est_method <string>: RSEM|kallisto|salmon
比如说我们RSEM来进行定量,相关命令如下:
$ TRINITY_HOME/util/align_and_estimate_abundance.pl --transcript Trinity.fasta --est_method RSEM --aln_method bowtie --seqType fq --left reads_1.fq --right reads_2.fq --trinity_mode --prep_reference --output_dir rsem_outdir之后会返还两个输出文件:
’RSEM.isoforms.results’、’RSEM.genes.results’基因分型的结果如下所示:
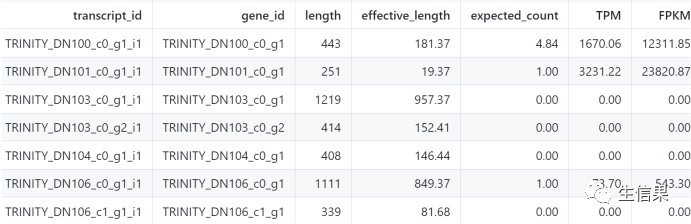
每一行对应了一个转录本,记录了转录本的长度,有效长度,期望数量,TPM和FPKM,可以看到第二行这个基因有着最高的表达量。
至此,我们可以对每个样本构建转录本和基因的表达矩阵,使用下面这个脚本:
$ TRINITY_HOME/util/abundance_estimates_to_matrix.pl# --est_method <string>: RSEM|kallisto|salmon# --gene_trans_map <string>: 基因和转录本的转换文件
例子如下:
$ TRINITY_HOME/util/abundance_estimates_to_matrix.pl --est_method RSEM --gene_trans_map Trinity.fasta.gene_trans_map --name_sample_by_basedir我们会得到以下三个文件:
‘RSEM.isoform.count.matrix’: 原始counts数据
‘RSEM.isoform.TPM.not_cross_norm’: TPM表达量(没有进行跨样本归一化)
‘RSEM.isoform.TMM.EXPR.matrix’: TMM归一化的表达值
2.差异表达分析
得到了表达矩阵之后我们就可以进行差异表达分析了,Trinity中使用的是EdgeR这个R包来做差异表达分析,同样需要咱们的系统中装有相对应的R环境和软件包。
具体脚本命令和参数如下所示:
$ TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl# --matrix|m <string>: 原始count矩阵# --method <string>: edgeR|DESeq2|voom
运行以下脚本:
$ TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl --matrix RSEM.isoform.count.matrix --method edgeR –samples_file samples_described.txt关于edgeR的参数设置可以查询edgeR manual。
然后我们会得到三个文件,分别是:
‘${prefix}.sampleA_vs_sampleB.${method}.DE_results’
‘${prefix}.sampleA_vs_sampleB.${method}.MA_n_Volcano.pdf’
${prefix}.sampleA_vs_sampleB.${method}.Rscript’
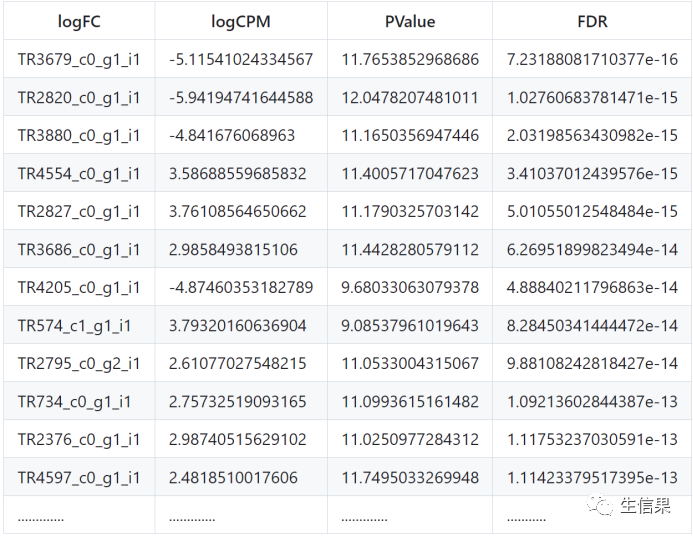
我们一起来看看DE_results文件的头几行,记录着log fold change和相对应的统计量(Pvalue,FDR矫正)
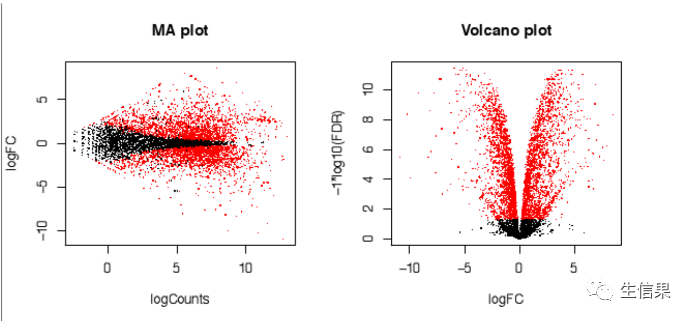
同时还有火山图
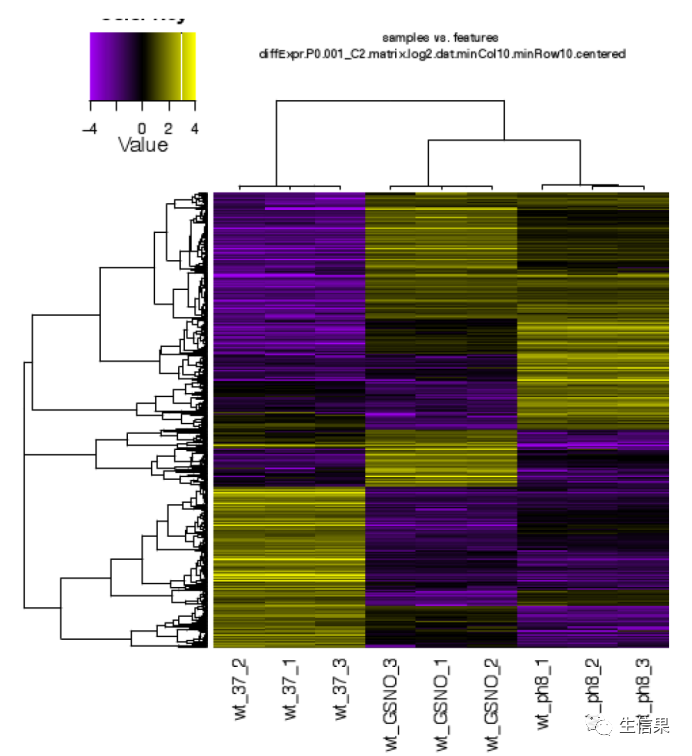
对于差异表达的一个比较初始的分析就是去获取那些前后表达差异极大的转录本(最显著的FDR和fold–change),然后根据不同的表达模式将这些转录本划分聚类。我们可以使用以下脚本进行聚类
TRINITY_HOME/Analysis/DifferentialExpression/analyze_diff_expr.pl--matrix <string>: TMM.EXPR.matrix-P <float>: pvalue阈值-C <float>: 最小的fold change数
如小果下面举的例子:
TRINITY_HOME/Analysis/DifferentialExpression/analyze_diff_expr.pl --matrix ../Trinity_trans.TMM .EXPR.matrix -P 1e-3 -C 2然后我们就能得到如下差异基因和不同样本的热图:
3.转录本功能注释
在我们得到了拼接的转录本以及相应的基因集之后,我们自然而然想知道这些基因富集在什么样的功能上。同样的,咱们强大的Trinity提供了一套完整的流程。首先我们通过以下脚本来获取每个基因所对应的GO term:
TRINITY_HOME/util/extract_GO_assignments_from_Trinotate_xls.pl --Trinoate_xls trinotate.xls -G --include_ancestral_terms > go_annotations.txt然后我们再使用Bioconductor中的包GOseq进行功能富集:
TRINITY_HOME/Analysis/DifferentialExpression/run_GOseq.pl --factor_labeling factor_labeling.txt --GO_assignments go_annotations.txt --lengths gene.lengths.txt
其中’factor_labeling.txt’这个文件记载着不同样本间的差异表达基因集,如下图所示:

而对于‘gene.lengths.txt’这个文件,我们使用如下命令进行创建:
TRINITY_HOME/util/misc/fasta_seq_length.pl Trinity.fasta > Trinity.fasta.seq_lengthsTRINITY_HOME/util/misc/TPM_weighted_gene_length.py --gene_trans_map trinity_out_dir/Trinity.fasta.gene_trans_map --trans_lengths Trinity.fasta.seq_lens --TPM_matrix isoforms.TMM.EXPR.matrix > Trinity.gene_lengths.txt
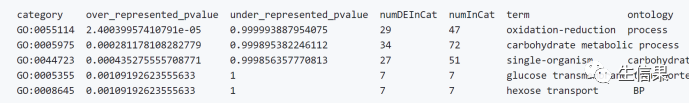
在最后我们就会得到一个如下的图标,其中记录着差异表达基因富集到的功能通路:
以上就是使用Trinity进行de novo转录本拼接、下游分析的全部内容,如果你有什么问题的话欢迎和小果讨论!
如果小伙伴有其他数据分析需求,可以尝试使用本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:(http://www.biocloudservice.com/home.html),其中也包括了通路表达分析(http://www.biocloudservice.com/313/313.php),单细胞的基因共表达分析(http://www.biocloudservice.com/906/906.php)等各种小工具哦~,有兴趣的小伙伴可以登录网站进行了解。
点击“阅读原文”立刻拥有
↓↓↓