强大的bam文件处理工具——sambamba

小果最近在做bam文件的处理,samtoools虽然功能很强大,但文件太大的话还是很耗时的,而且在给bem文件构建索引时还出现了下面的错误/(ㄒoㄒ)/~~
$ samtools index -b test.sort.bam test.sort.bam.bai[E::hts_idx_check_range] Region 536870922..536871063 cannot be stored in a bai index. Try using a csi index[E::sam_index] Read 'E00548:269:HV7NVCCXY:3:2117:26494:57301' with ref_name='chr1H', ref_length=558535432, flags=81, pos=536870923 cannot be indexedsamtools index: failed to create index for "Atlas.sort.bam": Numerical result out of range
错误信息表明区域“536870922..536871063”无法存储在“.bai”索引中,并建议尝试使用“.csi”索引。这是因为“.bai”索引格式在可以处理的最大参考序列长度方面存在限制,即“229-1”或536870911。如果参考序列长度超过此值,则需要使用.csi索引格式。
但是!但是GATK等一些生信工具似乎不支持“.csi”格式的索引文件,怎么办!(;´༎ຶД༎ຶ`)还好,小果找到了sambamba,它不仅能够给最大参考序列长度的bam文件构建bai索引,而且它还能快速、高效、准确地分析你的BAM文件,它能利用多核处理器,这也就意味着大大的提高了文件处理效率。
sambamba是什么?
sambamba是一个新的BAM文件处理工具,它使用了D语言的多线程和异步IO特性,实现了高效的并行化处理。sambamba可以在多核CPU上同时运行多个任务,利用硬盘和内存的带宽,提高了处理速度。sambamba还使用了一些优化算法和数据结构,比如快速排序,哈希表,位图等,减少了内存占用和磁盘读写。sambamba支持了samtools和picard的大部分功能,而且速度更快,内存占用更少,操作更简单。sambamba不仅可以对BAM文件进行排序、索引、过滤、统计、标记重复等常见的操作,还可以进行一些特殊的功能,比如区域过滤,标记重复序列,检测结构变异等。sambamba还支持多种输入和输出格式,比如CRAM、SAM、BED、VCF等,让你可以灵活地处理各种数据类型。
下载网址:https://github.com/biod/sambamba/releases
可以通过以下方法下载并安装:
git clone --recursive https://github.com/lomereiter/sambamba.gitcd sambamba && make sambamba-ldmd2-64
输入以下命令,若出现版本信息,则安装成功!
sambamba –version
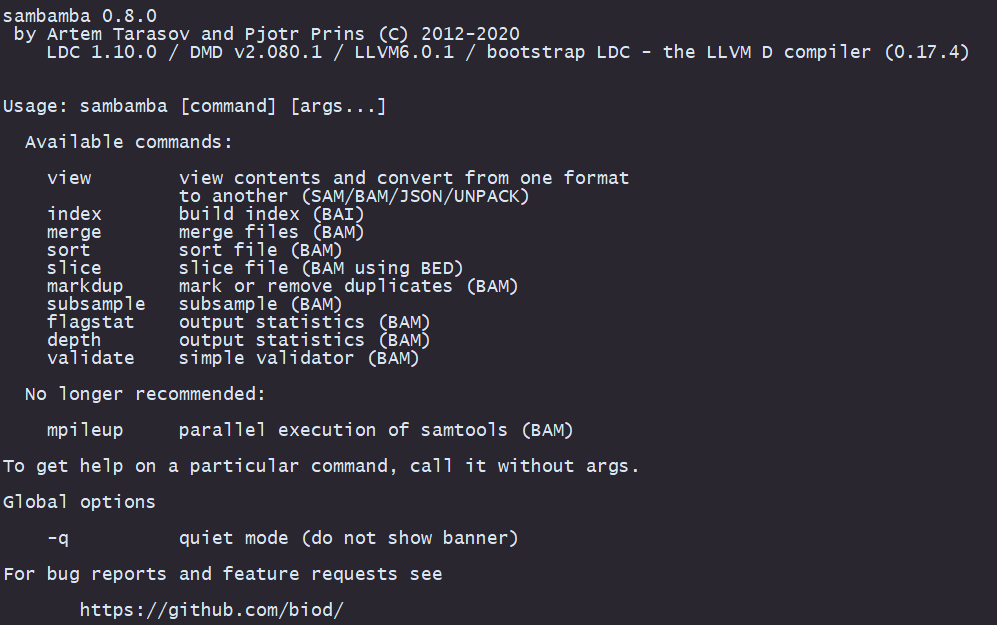
看一下sambambad 功能吧!Sambamba的主要功能如下:
sort: 对SAM/BAM文件进行排序,可以按照位置或者名称排序,支持多线程和压缩。
index: 对BAM文件建立索引,可以加速后续的查看和切片操作。
view: 查看SAM/BAM文件的内容,可以指定输出格式和过滤条件,也可以查看参考序列的信息。
merge: 合并多个BAM文件,可以自动处理header和压缩级别。
flagstat: 统计BAM文件中的reads的标志位,可以显示QC通过和失败的reads数目,以及各种配对情况和重复情况。
markdup: 标记或者移除BAM文件中的重复reads,可以设置临时文件目录和压缩级别。
slice: 提取BAM文件中的某个区域,可以指定输出格式和过滤条件。
subsample:对BAM文件进行子采样。子采样是指从原始数据中随机选择一部分数据,以便在保留原始数据特征的同时减少数据量,可以提高计算效率。
depth 输出统计信息(BAM)
validate 简单验证器(BAM)
不再支持变异检测(可以用bcftools)
对于上面构建索引时出现的的错误,用sambamba再尝试以下:
sambamba index test.sort.bam test.sort.bam.bainice!成功啦!而且我的文件是41GB,仅用了两分钟就完成了!

这只是一个构建索引的例子,sambamba还可以做很多其他有用的事情。或者,你可以用它来转换文件格式,统计BAM文件中的覆盖度、深度、映射质量等信息。
情景一:SAM转BAM
Sambamba 的view命令可以用来查看SAM/BAM格式文件的内容并进行格式转换。它提供了许多选项来控制输出,包括:
·-F, –filter=FILTER:设置对齐的自定义过滤器。
·–num-filter=NUMFILTER:过滤标志位;’i1/i2’对应于samtools的-f i1 -F i2参数;两个数字都可以省略。
·-f, –format=sam|bam|json|unpack:指定输出格式(默认为SAM);unpack流解压BAM。
·-h, –with-header:在读取前打印标题(BAM输出始终如此)。
·-H, –header:仅将标题输出到stdout(如果format=bam,则标题以SAM格式打印)。
·-I, –reference-info:仅将参考名称和长度以JSON格式输出到stdout。
·-L, –regions=FILENAME:仅输出与BED文件中的一个区域重叠的读取。
·-c, –count:仅将匹配记录数输出到stdout,hHI被忽略。
·-v, –valid:仅输出有效对齐。
·-S, –sam-input:指定输入为SAM格式。
·-T, –ref-filename=FASTA:指定写入参考(NA)。
·-p, –show-progress:在STDERR中显示进度条(仅适用于未指定区域的BAM文件)。
·-l, –compression-level:指定压缩级别(从0到9,仅适用于BAM输出)。
·-o, –output-filename:指定输出文件名。
·-t, –nthreads=NTHREADS:使用的最大线程数。
·-s, –subsample=FRACTION:子采样读取(读取对)。
·–subsampling-seed=SEED:设置子采样种子。
例:sambamba view -S -f bam -p input.sam -o output.bam
在上面的命令中,-S选项指定输入文件为SAM格式,-f bam选项指定输出格式为BAM,-p选项指定显示进度条,input.sam是输入文件的名称,而-o output.bam指定输出文件的名称。
情景二:统计每个染色体上的覆盖度
你可以使用以下命令:
sambamba depth base test.bam -o coverage.txt这样就会生成一个文本文件,列出了每个染色体上每个碱基位置的覆盖度。你可以用类似的方式统计其他信息,比如区域覆盖度、基因覆盖度、映射质量分布等。
如果你仅某段基因组感兴趣,想要提取比对到这段区域的bam信息,那就用slice试试吧,命令如下:
slice input.bam chr1H:10000-20000 > output.bam这样我们就得到我们的靶区文件啦!
甚至可以用它来标记重复序列,并且选择保留或删除这些重复序列。假设你想要标记test.bam文件中的重复序列,并且删除它们,你可以使用以下命令:
sambamba markdup -r test.bam dedup.bam-r 表示删除重复,默认仅标记不删除。这样就会生成一个新的BAM文件,去除了所有被标记为重复的reads。你也可以选择保留重复序列,但是给它们添加一个标签,以便于后续分析。
总之,sambamba是一个功能强大、性能优异、易于使用的BAM文件处理工具,它可以让你的生物信息学分析更加高效和愉快。如果你还没有尝试过sambamba,那么赶快去下载吧!
往期推荐
