小果发现用SRA Toolkit工具下载转录组数据很好用!
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

有时候我们想要下载别人的转录组数据进行练习或分析,可能会突然发现无从下手,巧妇难为无米之炊啊,这可怎么办?
小果本期来分享用SRA Toolkit工具在NCBI的SRA数据库下载拟南芥的转录组数据Run Selector :: NCBI (nih.gov)。
SRA Toolkit是由美国国家生物技术信息中心(NCBI)开发的一款用于高通量测序数据处理的软件包,主要用于存储和分析NCBI Sequence Read Archive(SRA)中的测序数据。该工具包提供了多个命令行工具,支持从SRA下载数据、将SRA转换为常见的FASTQ格式、将FASTQ文件转换为SRA格式等多种功能。
SRA Toolkit不仅适用于NCBI SRA数据库,也可用于处理其他来源的高通量测序数据。使用SRA Toolkit可以实现高效的数据下载、数据格式转换、数据处理和分析等多个步骤,并且支持多线程和分布式处理,能够加快数据处理速度和提高数据处理效率。下面我们一起来看看这个工具如何使用吧~
1. 安装SRA Toolkit
conda create --name sratoolkitconda activate sratoolkitconda install -c bioconda sra-tools
2.下载数据ls
激活sratollkit环境,然后就可以用它批量下载数据了,因为数据量太大,而小果只是想作为练习,因此只下载了9个 样本的测序数据,具体方法如下:
(1)查找需要下载的SRA数据的访问号(Accession Number)
(2)下载Accession List,会下载一个包含所有Run编号的文本文档
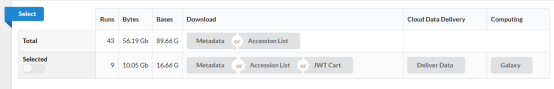

小果这里只保留了9个样本(在文档里删除其他不需要的run编号就可以了),需要的话可以全部下载。
(3)在终端中切换到要将SRA文件下载到的目录
nohup prefetch -O . $(<srr_acc_list.txt) &<=”” span=””>###将文件下载到当前目录并作为后台进程运行
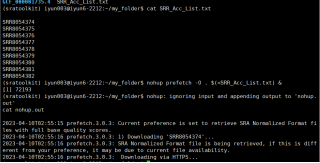
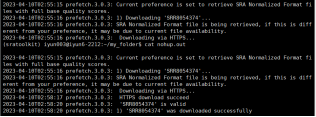
当然了,你也可以这样下载,不过会比较麻烦:
prefetch SRR8054374 SRR8054375 SRR8054376 SRR8054377 SRR8054378 SRR8054379 SRR8054380 SRR8054381 SRR8054382### SRR8054374等为SRA文件的run numbera year later……
好啦,可以看到文件已经下载完成了。
3.转换格式
如果你下载的文件在同一个目录下:
fastq-dump --gzip --split-files *.sra &#将.sra文件转换为.fastq.gz文件gunzip *.fastq.gz #解压缩所有.fastq.gz文件
以上就是全部内容啦,我们下期见!
欢迎使用:云生信 – 学生物信息学 (biocloudservice.com)
如果想用服务器可以联系微信:18502195490(快来联系我们使用吧!)

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓