UMAP:生物信息分析的新宠,降维神器解析高维数据!

@
点击蓝字
关注我们
生物信息分析的过程中,我们常常会碰到许多维度很高的数据集,这个时候我们就需要用到降维技术了。今天小师妹就给大家带来一个降维的新方法——UMAP
UMAP (Uniform Manifold Approximation and Projection) 是一种非线性降维技术,用于将高维数据映射到低维空间。它可以用于可视化数据集或降低数据维度,将高维数据映射到低维空间,以便进行可视化和进一步的分析。实际上,UMAP 可以被看作是 t-SNE (t-distributed Stochastic Neighbor Embedding) 的一种改进版本,它使用了一些新的数学技术来提高效率和可扩展性。UMAP 的核心思想是通过在数据流形结构上建立连通性图,来保留原始数据的局部和全局结构。UMAP 首先使用随机梯度下降法来生成连通性图,然后将图中的节点映射到低维空间中。这个过程中,UMAP 使用了一些新的数学技术,包括 Riemannian geometry、algebraic topology 和 spectral graph theory 等,以提高算法的效率和可扩展性。
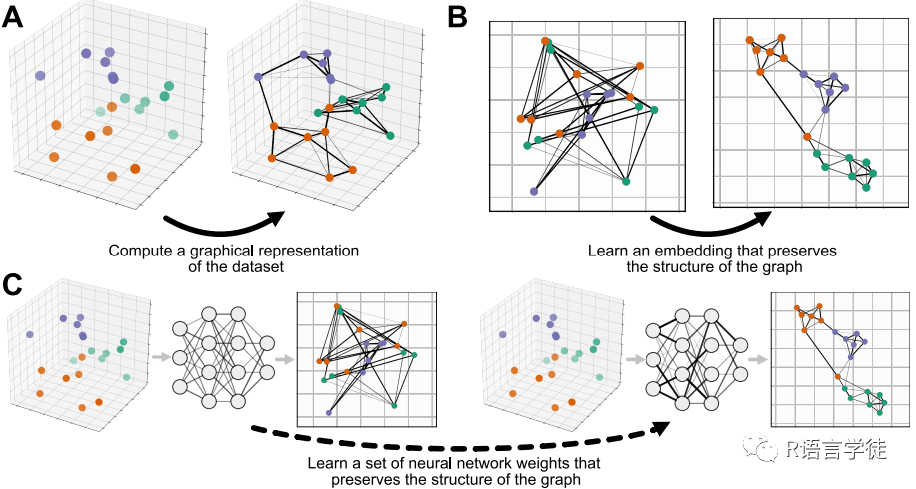

那么,我们要怎么在R语言中要怎么实现UMAP呢?这里小师妹给大家带来两个例子,我们首先会从简单的随机数据入手,接着以蛋白质表达序列数据为例,看看UMAP在生物信息分析的过程中的作用~
首先我们需要安装和加载UMAP包:
install.packages("umap")library(umap)
接下来,我们可以使用 umap() 函数来将数据降至二维空间,并进行可视化。
在这个示例中,我们生成了一个包含 10 个特征和 100 个观测值的随机数据集。
# 生成随机数据set.seed(123)data <- matrix(rnorm(1000), ncol = 10)
然后,我们使用 umap() 函数将数据降至二维空间,并将结果可视化。
# 进行 UMAP 降维embedding <- umap(data, n_components = 2)# 可视化结果plot(embedding$layout[,1], embedding$layout[,2], pch = 20)
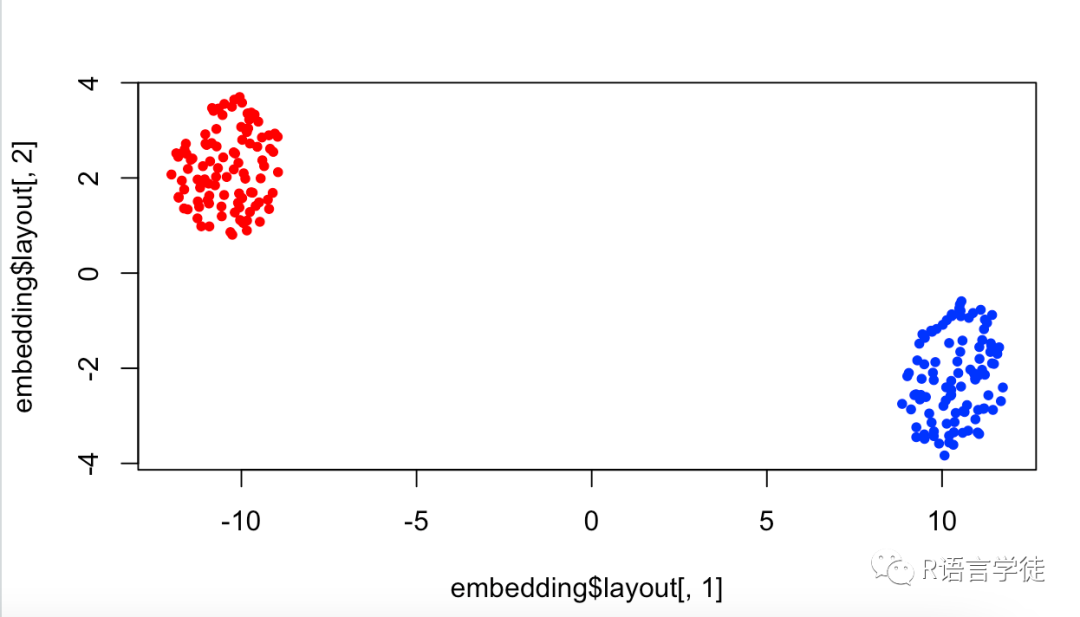
可以看到降维后的数据被很好地分成了两类
接下来我们看看实际生物数据的应用:
首先,导入数据,并且对数据进行预处理
data=read.csv(file.choose(),header = TRUE)data <- na.omit(data)X <- data[,2:79]# 为data设置新列X$Genotype <- ifelse(X$Genotype == "Control", 1, 0)
接着就可以直接使用umap函数进行 UMAP 降维啦
embedding <- umap(X, n_components = 2)这里,我们还可以根据 Genotype 列为每个观测值分配颜色值,使得最后可视化对结果更清晰
colors <- ifelse(X$Genotype == 1, "red", "blue")最后就可以得到可视化的结果
plot(embedding$layout[,1], embedding$layout[,2], col = colors, pch = 20)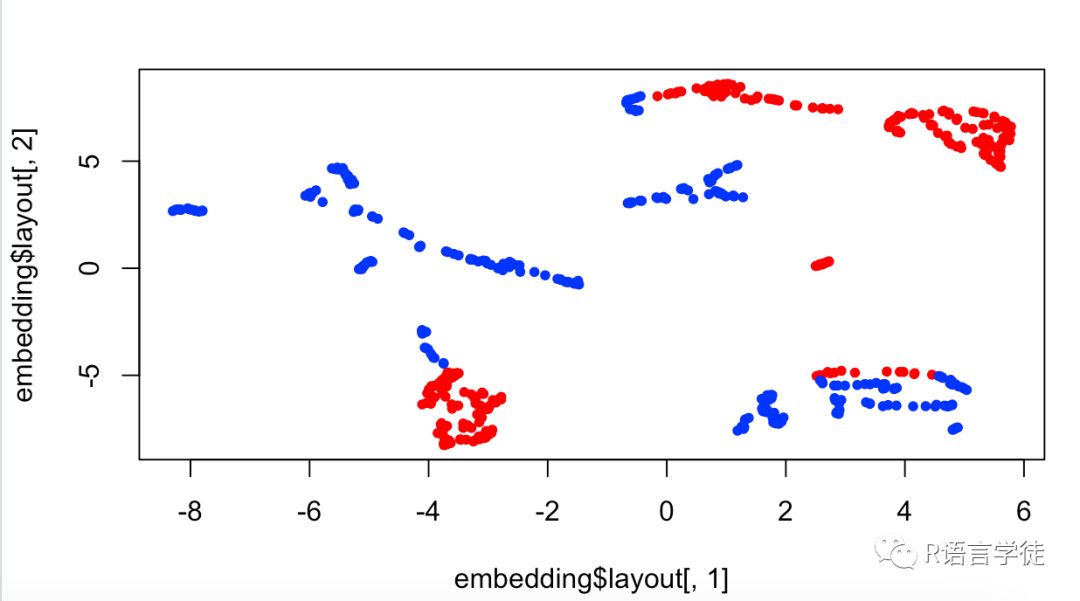
在生物信息的研究过程中,高维数据是一道无法避免的难关,而UMAP 是一种非线性降维技术,可以将高维数据映射到低维空间。UMAP 具有许多优点,包括处理大型数据集、保留原始数据的局部和全局结构、适用于各种类型的数据和自适应处理不同尺度的数据等。在 R 语言中使用 UMAP 也非常简单,可以直接使用 umap( ) 函数进行降维和可视化。UMAP 的应用可以帮助大家更好地理解和分析高维生物数据。这就是今天小师妹给大家带来的内容啦~大家学会了吗~
更多方便实用的小工具在云生信平台等着大家哦!
http://www.biocloudservice.com/home.html

END

