这也可以实现吗?单细胞基因共表达分析
{ 点击蓝字,关注我们 }
叮咚!今天又到小师妹分享的时间啦!今天小师妹又给大家带来了一个新的分析思路,是什么思路呢?展示单细胞基因共表达,以及进行共表达基因相关性分析,小伙伴是不是觉得有点意思呀!那还等着干嘛!赶紧跟着小师妹开启今天的学习之旅吧!
如何进行单细胞基因共表达分析?
单细胞基因共表达分析?小师妹以前好像没有在单细胞数据中听到过该分析,有可能是小师妹学艺不精奥!哈哈哈哈哈!我相信跟着小师妹一起学习之后,小伙伴们就会觉得很简单啦!接下来就让小师妹为大家讲解一下该如何分析?首先需要完成单细胞数据常规分析,然后直接利用FeaturePlot函数来绘制UMAP图上共表达基因可视化,可以选择自己感兴趣的两个基因来展示,很方便,其实,最重要的是小伙伴要掌握单细胞分析流程,这是基础奥!进行基因共表达可视化很简单,正如小师妹所说,如果觉得有点怀疑,那就跟着小师妹开始今天的实操吧!
准备需要的R包
library(Seurat)library(ggplot2)library(ggpubr)library(patchwork)library(magrittr)
数据准备
数据为标准的10x单细胞数据,将数据保存在一个文件夹中,包括barcodes.tsv,genes.tsv和matrix.mtx三个文件。
scRNA常规分析流程
#读取10x单细胞数据,需要提供数据所在文件夹路径pbmc.data <- Read10X(data.dir = hg19)#建立单细胞对象pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)# 计算线粒体基因比例pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")# 通过控制基因数和线粒体基因比例过滤掉低质量细胞pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)# 进行normalize并计算高变异基因pbmc <- NormalizeData(pbmc)pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)# Scale数据all.genes <- rownames(pbmc)pbmc <- ScaleData(pbmc, features = all.genes)# PCApbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))# 选择前10个pc基因降维,采用UMAP方式pbmc <- FindNeighbors(pbmc, dims = 1:10)pbmc <- FindClusters(pbmc, resolution = 0.5)pbmc <- RunUMAP(pbmc, dims = 1:10)#手动添加注释new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet")names(new.cluster.ids) <- levels(pbmc)pbmc <- RenameIdents(pbmc, new.cluster.ids)pbmc[['cell_type']] <- pbmc@active.identlevels(pbmc)
FeaturePlot绘制UMAP图上共表达基因展示,blend设置为TRUE
FeaturePlot(pbmc, features = c("CD3G", "CD247"),blend = TRUE,cols = c("gray80","red", "green"),pt.size = 0.5, raster = F) +theme(aspect.ratio = 1)ggsave("UMAP_co-expression_matrix.pdf", width = 12, height = 4)
自定义函数计算共表达基因相关性并可视化
#object对seurat对象,gene1和gene2为两个共表达基因名,cor.method为计算各相关性方法,可选spearman或pearson# 为了防止所有数据聚集在一起,这是jitter.num使数据进行轻微抖动,设置为0则为原始数据绘图# pos若为TRUE,怎只使用两个基因都表达的细胞计算相关性,可避免好多细胞中对其中一个细胞表达量为0getScatterplot <- function(object, gene1, gene2, cor.method = "spearman",jitter.num = 0.15, pos = TRUE){if (!gene1 %in% rownames(object)) {print("gene1 was not found")if (!gene2 %in% rownames(object)) {print("gene2 was not found")}}else{exp.mat <- GetAssayData(object = object, assay = "RNA") %>% .[c(gene1,gene2),] %>%as.matrix() %>% t() %>% as.data.frame()if (pos) {exp.mat <- exp.mat[which(exp.mat[,1] > 0 & exp.mat[,2] > 0),]}plots <- ggplot(data=exp.mat, mapping = aes_string(x = gene1, y = gene2)) +geom_smooth(method = 'lm', se = T, color='red', size=1)+stat_cor(method = cor.method)+geom_jitter(width = jitter.num, height = jitter.num, color = "black", size = 1, alpha=1)+theme_bw()+theme(panel.grid=element_blank(),legend.text=element_text(colour= 'black',size=10),axis.text= element_text(colour= 'black',size=10),axis.line= element_line(colour= 'black'),panel.border = element_rect(size = 1, linetype = "solid", colour = "black"),panel.background=element_rect(fill="white"))return(plots)}}#计算共表达基因相关性并可视化getScatterplot(pbmc3k, gene1 = "CD3G", gene2 = "CD247",jitter.num = 0.15, pos = TRUE) +theme(aspect.ratio = 1)ggsave("correlation_dotplot.pdf")
结果文件
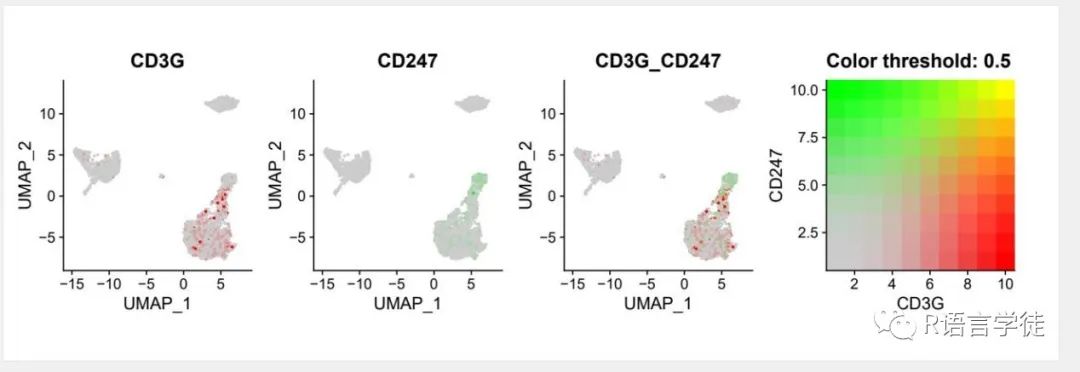
1.UMAP_co-expression_matrix.pdf
该结果图片为CD3G和CD247这两个T细胞marker基因进行基因共表达可视化。
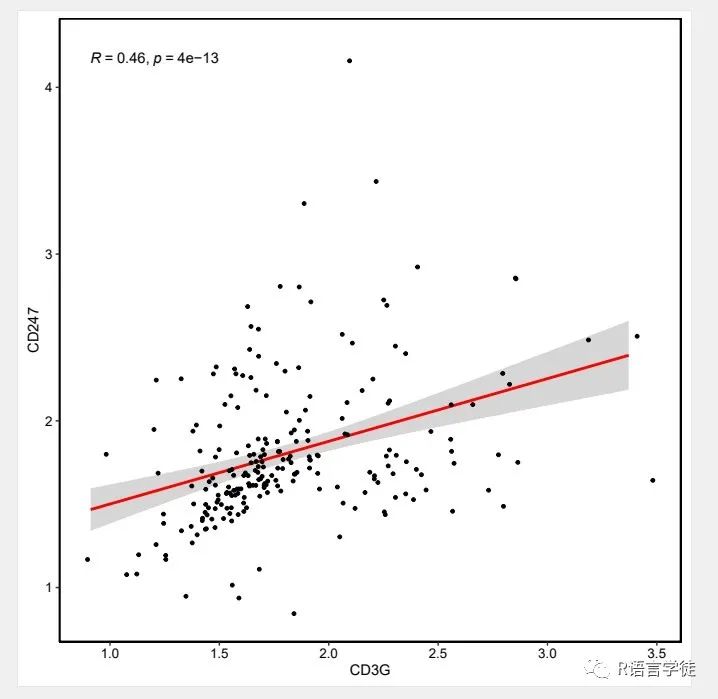
2.correlation_dotplot.pdf
该结果图片为共表达基因的相关性散点图。
今天小师妹对单细胞基因共表达的分享就到这里啦,进行该分析需要熟练掌握单细胞分析流程,单细胞相关分析内容可以尝试使用本公司新开发的云平台生物信息分析小工具,零代码完成分析,单细胞分析相关小工具,欢迎小伙伴来尝试哟!
云平台网址:
http://www.biocloudservice.com/home.html
单细胞分析:
http://www.biocloudservice.com/366/366.php
单细胞数据绘制小提琴图:
http://www.biocloudservice.com/788/788.php
绘制单细胞tSNE图:
http://www.biocloudservice.com/229/229.php
E
N
D

师妹微信
扫码添加