生信小白的福利,利用quanTIseq算法进行免疫浸润分析
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

小果今天给大家带来的分享绝对是生信热点分析内容,基于三种机器学习算法进行生存资料特征基因筛选,最终将三种算法获得的交叉基因作为我们的特征基因。
小果又来分享啦!今天小果为小伙伴带来的分享为利用quanTIseq算法进行免疫浸润分析,然后基于亚型分组信息文件,进行亚型间免疫细胞含量显著差异分析,该分析在肿瘤生信文章中必备的分析内容,非常值得小伙伴学习,小果强烈推荐呀!接下来跟着小果开启开始今天的分享学习。
1.quanTIseq算法介绍
在进行该分析之前,小果先简单介绍一下quanTIseq算法,quanTIseq基于反卷积算法,利用bulk samples的RNA_seq数据,可以对肿瘤样本中不同种类免疫细胞的组成进行预测,支持10种类型的免疫细胞,包括B cells,Classically activated macrophages (M1),Alternatively activated macrophages (M2),Monocytes,Neutrophils,Natural killer (NK) cells,Non-regulatory CD4+ T cells ,CD8+ T cells,Dendritic cells,Other uncharacterized cells。
这就是小果对该算法的介绍,想深入学习的小伙伴可以到该网址:https://icbi.i-med.ac.at/software/quantiseq/doc/index.html#Figure1
#首先下载quanTIseq流程分析脚本,如下文:wget https://icbi.i-med.ac.at/software/quantiseq/doc/downloads/quanTIseq_pipeline.sh#quanTIseq流程运行参数使用方法
2. 准备需要的R包
#安装需要的包install.packages("ggplot2")install.packages("ggsci")install.packages("tidyverse")install.packages("reshape2")install.packages("ggpubr")#加载需要的R包library(reshape2)library(ggpubr)library(ggplot2)library(tidyverse)library(ggsci)
3.输入数据
#EXP.txt,基因表达矩阵文件,行名为基因名,列名为样本信息。exp<-read.table("EXP.txt",header=T,row.names=1,sep="t",check.names=F)
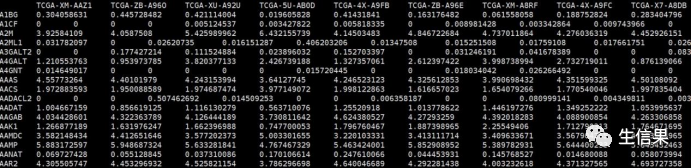
4.利用quanTIseq算法进行免疫浸润分析
#将行名转化为列名exp<-rownames_to_column(exp,var="GENE")4.利用quanTIseq算法进行免疫浸润分析#quanTIseq进行免疫浸润分析bash quanTIseq_pipeline.sh --inputfile=exp.txt --outputdir=quanTIseqTest --prefix=quanTIseqTest --pipelinestart=deconinput表示基因表达矩阵,第一列为基因名,其他列为样本名。outputdir表示生成结果文件名prefix表示生成结果文件前缀pipelinestart表示quanTIseq分析流程,基因表达矩阵选用decon#生成结果文件为quanTIseqTest_cell_fractions.txt,该结果为计算的10种免疫细胞在不同样本中的含量,第一列为样本名,其他列为免疫细胞类型。
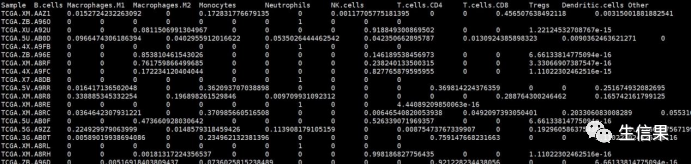
5.不同亚型中quanTIseq算法计算的免疫细胞的显著差异分析
#读取quanTIseq分析免疫细胞组成结果文件immune<-read.table("quanTIseqTest_cell_fractions.txt",header=T,sep="t")#将Sample列”.”修改为”-”immune$Sample<-str_replace_all(immune$Sample,"\.","-")#亚型分组文件,行名为样本信息,第一列为样本信息,第二列为亚型分组信息。K3<-read.table("K3.txt",header=T,sep="t")

final<-left_join(immune,K3,by=”Sample”)quanTIseq_cluster<-melt(final,id.vars=c("Sample","Cluster"))colnames(quanTIseq_cluster)<-c("Sample","Cluster","celltype","quanTIseq")boxplot_quanTIseq<- ggplot(quanTIseq_cluster, aes(x = celltype, y =quanTIseq ))+labs(y="quanTIseq",x= "")+geom_boxplot(aes(fill = Cluster),position=position_dodge(0.5),width=0.5)+scale_fill_npg()+theme_classic() +theme(axis.title = element_text(size = 12,color ="black"),axis.text = element_text(size= 12,color = "black"),panel.grid.minor.y = element_blank(),panel.grid.minor.x = element_blank(),axis.text.x = element_text(angle = 45, hjust = 1 ),panel.grid=element_blank(),legend.position = "top",legend.text = element_text(size= 12),legend.title= element_text(size= 12)) +stat_compare_means(aes(group = Cluster),label = "p.signif",method = "kruskal.test",hide.ns = F)ggsave(file="quanTIseq.pdf",boxplot_quanTIseq,height=10,width=15)
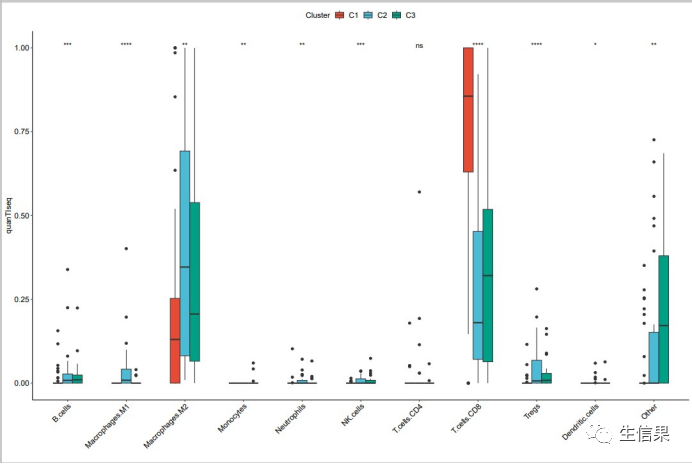
最终小果成功的对不同亚型利用quanTIseq算法进行了免疫浸润分析,,看起来图片效果非常不错,欢迎大家和小果一起讨论学习呀!免疫浸润相关分析内容,例如单样本富集算法分析免疫浸润丰度(http://www.biocloudservice.com/106/106.php),计算64种免疫细胞相对含量(http://www.biocloudservice.com/107/107.php),panImmune免疫浸润分析(http://www.biocloudservice.com/782/782.php)等都可以用本公司新开发的零代码云平台生信分析小工具,一键完成该分析奥,感兴趣的小伙伴欢迎来尝试奥,网址:http://www.biocloudservice.com/home.html。今天小果的分享就到这里,下期在见奥。
对了,如果大家还对其他生信的工具感兴趣的话,欢迎大家点击http://www.biocloudservice.com/home.html进行学习哦!
小果友情推荐
好用又免费的工具安利



点击“阅读原文”立刻拥有
↓↓↓