想要精准分析scRNASeq数据?M3Drop助你事半功倍!
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

-
M3Drop是一个强大的单细胞测序分析工具,那么你知道怎样通过M3Drop分析单细胞数据吗?感兴趣的话就和小果一起来看看吧!
什么是M3Drop?
M3Drop包是一个R包,主要用于单细胞转录组学(scRNASeq)数据的预处理和分析。它提供了用于估计噪声和dropout率的统计模型,以及用于识别差异表达基因的功能。主要作用是帮助用户快速准确地对单细胞转录组数据进行处理和分析,挖掘并识别出差异表达基因。那么我们就来一起看看它具体是怎么使用的吧!

数据准备与预处理
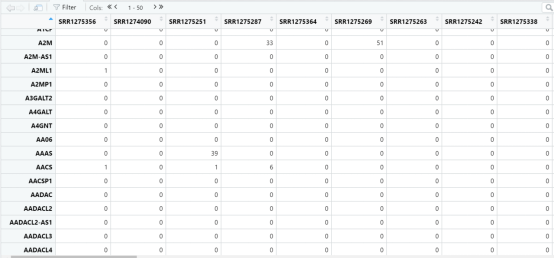
library(scRNAseq)data(fluidigm) #设置数据集fluidigm<-ReprocessedFluidigmData() #导入数据集ct <- floor(assays(fluidigm)$rsem_counts) #生成数据矩阵ct[1:4,1:4]counts <- ctsample_ann <- as.data.frame(colData(fluidigm))DT::datatable(sample_ann)View(ct)
我们先来看看我们导入的数据是什么样吧!

数据过滤
chose_gene= apply(counts,1,function(x) sum(x>0) )>0table(chose_gene)counts <- counts[chose_gene,]table((apply(counts,1,function(x) sum(x>0)>0 )))

M3Drop分析
通过上面的数据处理,我们进入今天的正题,那么M3Drop如何进行下一步的单细胞分析呢?和小果一起来看一下吧!
1.构建M3Drop对象
在使用M3Drop进行统计分析之前,我们首先使用M3DropCleanData()函数创建一个M3Drop对象。当我们使用该函数时,它将执行样本质量控制,并基于质量控制后的数据生成M3Drop对象。最终得到的对象将包含标签、数据等信息哦。
library(M3Drop)#生成M3Drop对象Normalized_data <- M3DropCleanData(counts,labels = sample_ann$Biological_Condition ,is.counts=TRUE,min_detected_genes=2000)
2.使用M3Drop统计分析
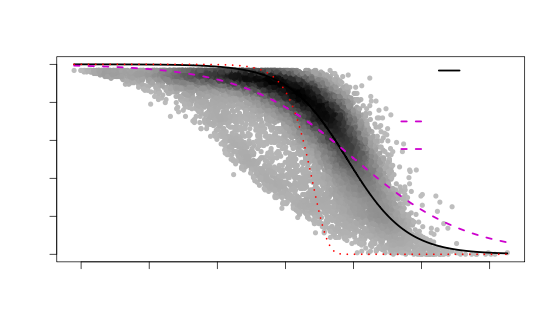
接下来,我们可以使用M3DropDropoutModels()函数执行M3Drop统计分析。该函数接收我们上面创建好的M3Drop对象,并返回MM、logistic和DoubleExpo模型的拟合。该函数的作用是使用统计模型来估计噪声和dropout率。
#使用M3Drop统计分析fits <- M3DropDropoutModels(Normalized_data$data)#计算每个模型的Sum absolute residualsdata.frame(MM=fits$MMFit$SAr,Logistic=fits$LogiFit$SAr,DoubleExpo=fits$ExpoFit$SAr)#计算每个模型的Sum squared residualsdata.frame(MM=fits$MMFit$SSr,Logistic=fits$LogiFit$SSr,DoubleExpo=fits$ExpoFit$SSr)
现在,我们一起来看看通过M3Drop分析后的效果图吧!
3.寻找和绘制差异基因
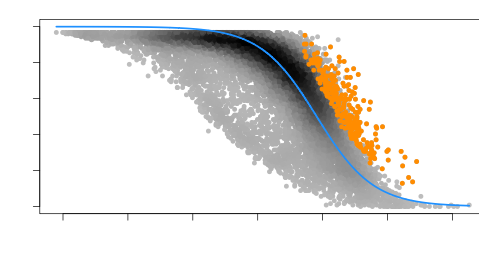
最后,我们可以使用M3DropFeatureSelection()函数找到差异表达的基因,并生成一个包含这些基因的数据框“DE_genes”。然后我们使用M3DropExpressionHeatmap()函数对差异表达基因进行热图绘制,以呈现其表达情况。
#找差异基因DE_genes <- M3DropFeatureSelection(Normalized_data$data,mt_method="fdr",mt_threshold=0.01)
现在我们一起来看看生成的差异基因模型图:
#绘制热图par(mar=c(1,1,1,1))heat_out <- M3DropExpressionHeatmap(DE_genes$Gene,Normalized_data$data,cell_labels = Normalized_data$labels)
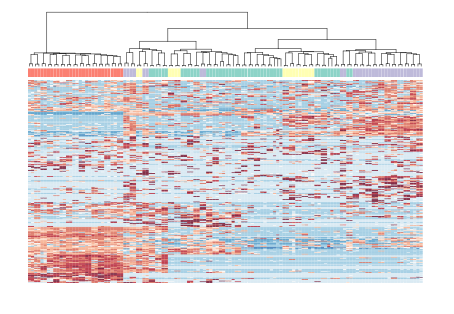
最后绘制好的差异基因表达热图
以上就是对scRNASeq数据进行M3Drop分析的完整过程,怎么样,你学会了吗?更多学习资源请大家多多关注小果云生信平台哦!

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓