GSEA富集分析——看了之后,小学三年级的妹妹都学会了!
点击蓝字
关注小图
今天小图总结了GSEA富集分析,先简单理解一下概念:
基因集富集分析(Gene Set Enrichment Analysis,GSEA)用一个预先定义的基因集中的基因来评估在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。
下面来看看软件具体操作
一
软件安装
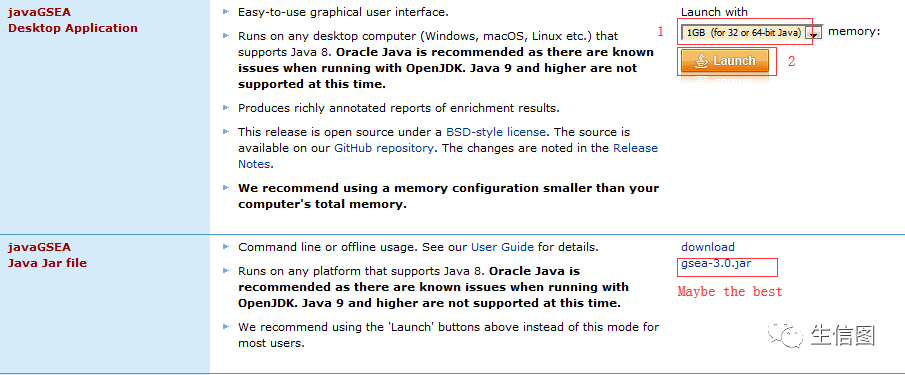
软件下载地址:http://software.broadinstitute.org/gsea/downloads.jsp
推荐使用官方的第一个软件,根据电脑系统和分析数据的大小可以选择下载不同相应版本的java。该软件是基于java环境运行的,而且需要联网。若会出现打不开的现象(小图就碰到了),要么是没有安装java,要么是java的版本不匹配,安装或相应版本的java就能打开。也可能是网络的问题,或java安全性问题,这时选择官网提供的第二个软件,同样依赖java运行,但不需联网,启动快。
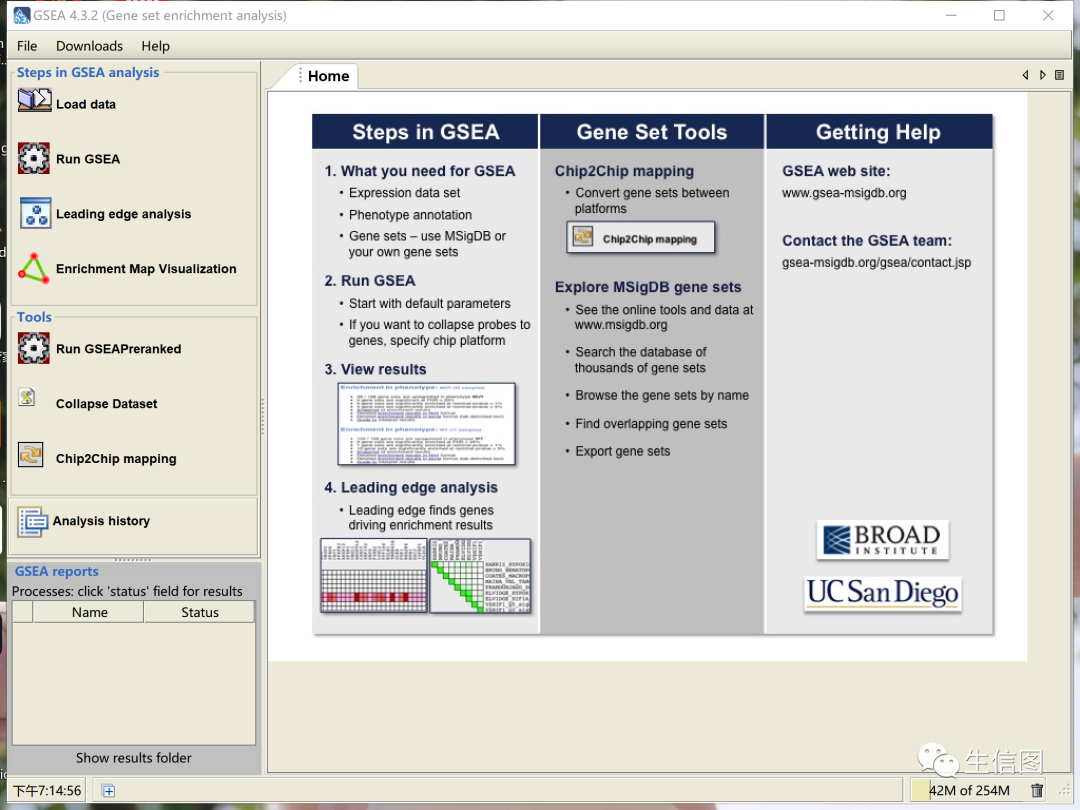
打开界面
二
数据准备
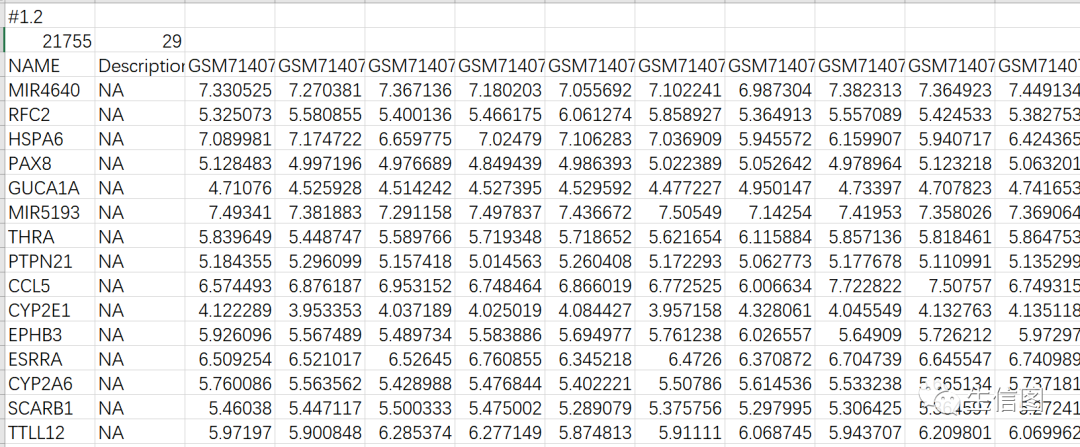
数据示例中两个文件都是表达矩阵,其中文件第一列是探针名字,文件的第一列是gene symbol。
第一行:,表示版本号,#1.2是固定格式;
第二行:两个数分别表示gene NAME的数量和样本数量(矩阵列数);
矩阵:第一列是gene NAME;第二列,没有的话可以全用NA;后面的就是基因在不同样本中标准化后的表达数据了。
接下来对以上文件进行保存,选择另存为,保存类型一定要选择文本文件(制表符分隔)(*.txt),在文件名里面先输入双引号,双引号里面填你需要的名字并加上.gct。比如小图的格式就是:”GE1_high_low_gct.gct”
样品分组信息
第一行:三个数分别表示:275个样品,2个分组,最后一个数字1是固定的;
第二行:以开始,键分割,分组信息(有几个分组便写几个,多个分组在比较分析时,后面需要选择待比较的任意2组);
第三行:样本对应的组名。样本分组信息的第三行,同一组内的不同重复一定要命名为相同的名字,可以是分组的名字。例如相同处理的不同重复在自己试验记录里一般是High_1、High_2、High_3,但是在这里一定都要写成一样的值。与表达矩阵的样品列按位置一一对应,名字相同的代表样品属于同一组。如果是样本分组信息,上图中的和也可以对应的写成和,更直观。但是,如果想把分组信息作为连续表型值对待,这里就只能提供数字。
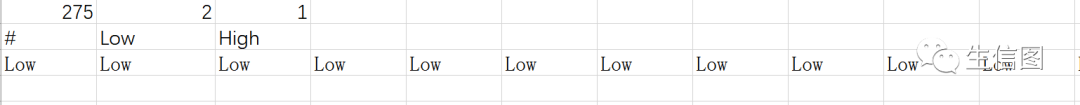
最后另存为,保存类型一定要选择文本文件(制表符分隔(*.txt),文件名里面先输入双引号,双引号里面填你需要的名字并加上.cls。比如小图的格式就是:”GE1_high_low_cls.cls”
三
分析参数设置和软件运行
演示使用的数据来自GSEA官网:
表达矩阵:Diabetes_collapsed_symbols.gct
样品分组信息:Diabetes.cls
数据导入
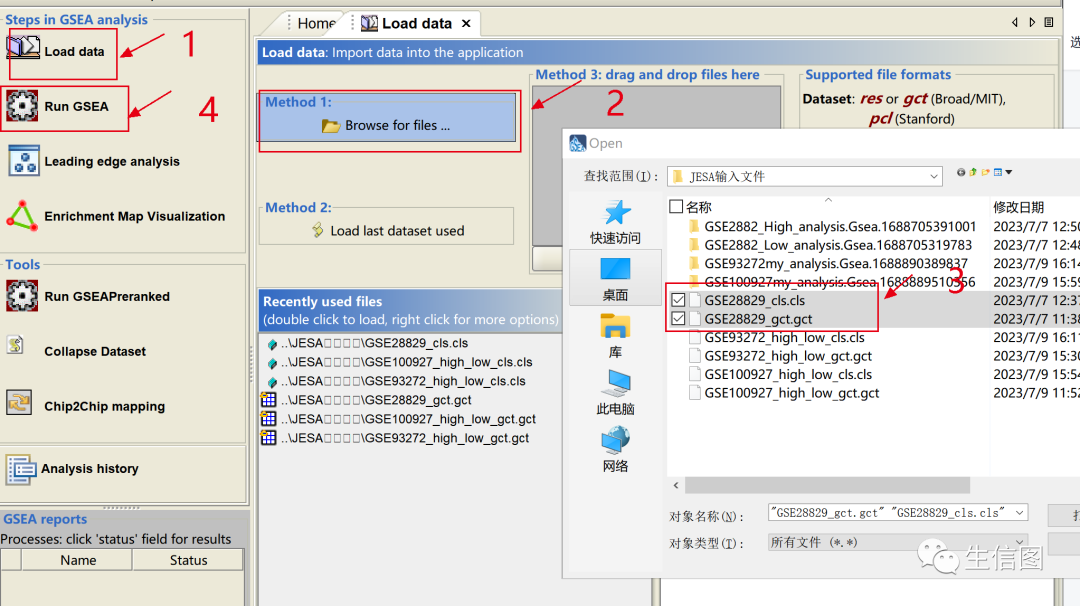
按照上图步骤依次点击——在弹出文件框中找到待导入的文件,选中点击即可;
若文件格式没问题会弹出一个提示的框,证明文件上传成功,并且会显示在所示的位置;若出错,请仔细核对文件格式。
注意:1)本地文件存放路径不要有中文、空格(用代替空格)和其他特殊字符;2)所有用到的文件都需要通过上述方式先上传至软件;3)数据上传错误后可以通过点击工具栏——进行清除。
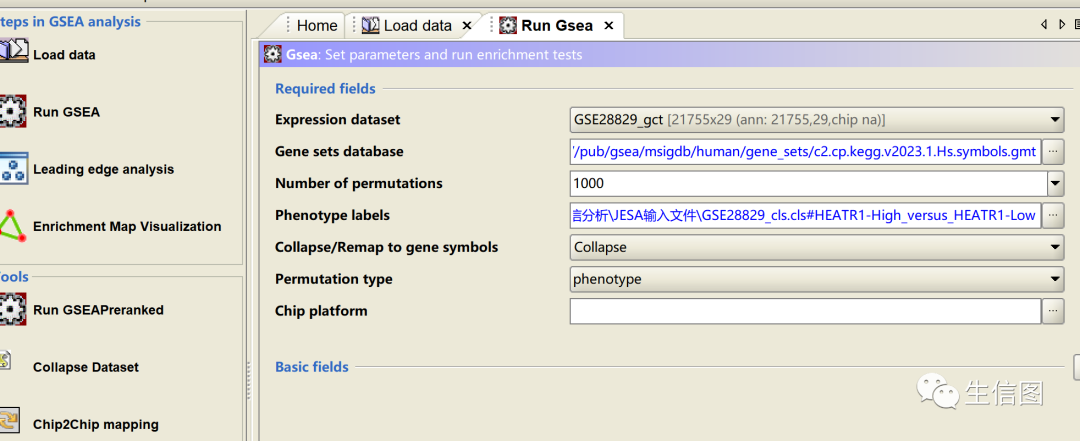
Expression dataset:导入表达数据集文件
Gene sets database:基因功能集数据库,如KEGG
Number of permutations:置换检验的次数,数字越大结果越准确,默认检验1000次。
Phenotype labels:选择比较方式,如果文件只有2个组别的话,任意选一个就行,哪个在前在后全在自己怎么解释方便;如果数据有多组的话,GSEA会提供两两间比较的组合选项或者某一组与剩下所有组的比较。
Collapse dataset to gene symbols:如果表达数据集文件中NAME已经与gene sets database中名字一致,选择No_Collapse,反之选择Collapse。
Permutation type:每组样本数目大于7个时,建议选择phenotype,否则选择gene sets。
Chip platform:表达数据集为芯片数据时才需要,目的是对ID进行注释转换,如果已经转换好了就不需要了。应该也适用于其它需要转换ID的情况,不过事先转换最方便(在上次小图分享ID如何转换)。
基本设置
具体设置见下图

通常选择默认参数即可,在此简单介绍一下
Analysis name:取名需要注意不能有空格。如果做的分析多,最好选择一个有意义的名字,比如 (生信图),方便查找。
Enrichment statistic:基因集富集分析的最后一部分给出了GSEA中所用方法的数学描述。
Metric for ranking genes:基因排序的度量
Gene list sorting mode:对表达数据集中的基因进行排序,默认或者绝对值排序;
Gene list ordering mode:使用此参数确定表达数据集中基因是按照降序(默认)或者升序排列;
Max size & Min size:从功能基因集中筛选出不属于表达数据集中的基因后,剩下基因总数在此范围内则保留下来做后续的分析,否则将此基因集排除;一般太多或太少都没有分析意义。
Save results in this folder:在此可以选择分析文件在本地电脑的存储地址。
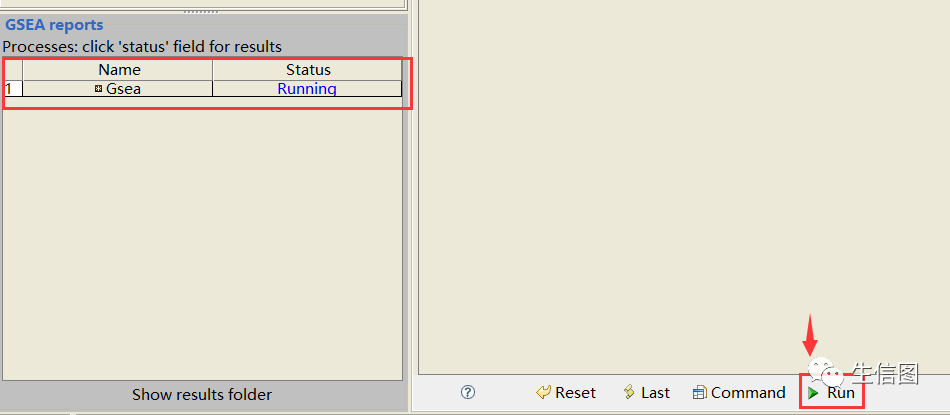
以上参数都设置好后点击参数设置栏下方的一个绿色按钮,若软件左下方处的状态显示的话则表示运行成功。
好了,小图今天的分享就到这结束啦,换上自己的数据趁热赶紧试一下吧!希望对大家的科研工作有所帮助。有兴趣的朋友可以继续关注小图的微信公众号(生信图)和云生信生物信息学平台( http://www.biocloudservice.com/home.html)。我们下期再见~
欢迎使用:云生信平台 ( http://www.biocloudservice.com/home.html)

|
往期推荐 |
|
最好懂的GEO数据下载和预处理,你要看吗? |
|
一文解决GEO芯片数据探针ID转换的工作! |
|
作为生信小白,怎样用做NGS的流程分析? |
👇点击阅读原文进入网址