让你的数据“开口说话”!从统计到可视化,把热图、PCA与箱线图一网打尽!

大家好,我是小果~在生物科学的海洋中,基因表达数据如同复杂的密码,等待我们去解读。今天小果将带小伙伴们一起探索基因数据集处理过程,让数据可视化成为我们的“翻译官”!从直观的热图展示基因活跃度,到主成分分析(PCA)揭示数据的深层结构,再到箱线图揭示基因表达值的分布规律,每一步都让我们更深入地理解基因世界的奥秘。无需复杂的数学公式,只需掌握这些可视化工具,我们就能让数据“开口说话”,轻松解读基因表达背后的故事。快来跟随我们的步伐,一起探索基因表达的无限可能吧!同时呢,十年的生信之路,小果已经练就了一身扎实的本领,现在准备用这身本事为小伙伴们服务啦!如果你在生信分析上遇到了难题,那就来找小果吧!小果会用自己的专业知识和技能,为你解决困扰,助你一臂之力。期待你的联系哦~
一、用到的R包简要介绍
二、R语言代码实操
公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240520-2
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

if (!requireNamespace("Biobase", quietly = TRUE)) {install.packages("Biobase")}library(Biobase)
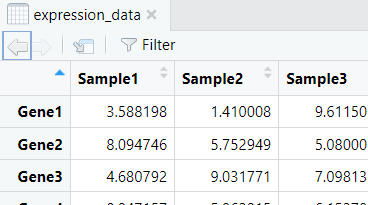
genes <- c("Gene1", "Gene2", "Gene3", "Gene4", "Gene5")samples <- c("Sample1", "Sample2", "Sample3")set.seed(123)expression_data <- matrix(runif(15, min = 1, max = 10), nrow = 5, ncol = 3,dimnames = list(genes, samples))
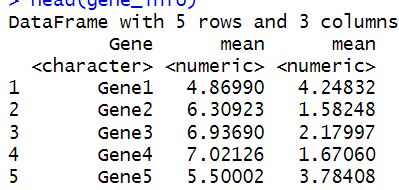
gene_stats <- apply(expression_data, 1, function(x) c(mean = mean(x), sd = sd(x)))gene_info <- DataFrame(Gene = genes, t(gene_stats))colnames(gene_info)[-1:-2] <- c("mean", "sd")head(gene_info)
if (!requireNamespace("MASS", quietly = TRUE))install.packages("MASS")library(MASS)
plot(expression_data, main = "Gene Expression Scatterplot Matrix")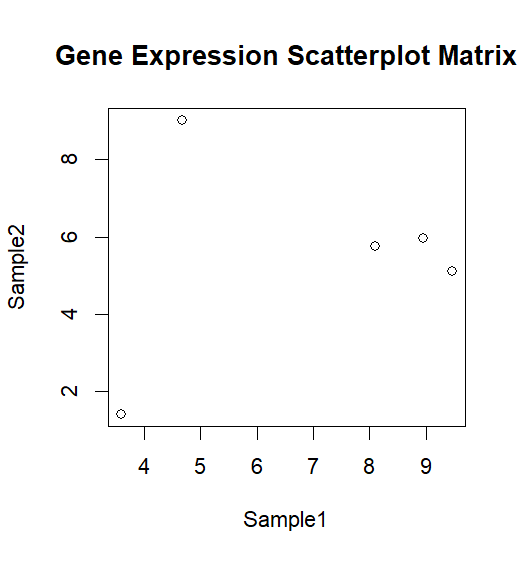
信息添加到gene_info数据框中。normalized_data <- t(scale(t(expression_data)))gene_info <- cbind(gene_info, DataFrame(normalized_mean = rowMeans(normalized_data), normalized_sd = apply(normalized_data, 1, sd)))colnames(gene_info)[5:6] <- c("normalized_mean", "normalized_sd")head(gene_info)

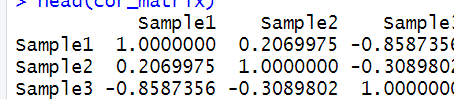
cor_matrix <- cor(expression_data, method = "pearson")head(cor_matrix)
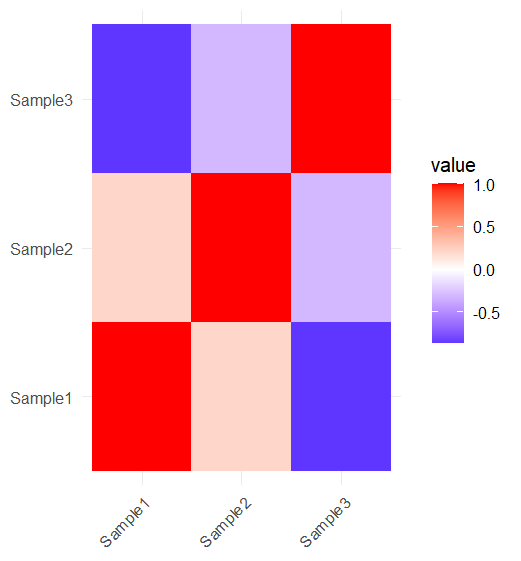
cor_melt <- reshape2::melt(cor_matrix)ggplot(cor_melt, aes(Var1, Var2, fill = value)) +geom_tile() +scale_fill_gradient2(low = "blue", mid = "white", high = "red", midpoint = 0) +theme_minimal() +theme(axis.text.x = element_text(angle = 45, hjust = 1),axis.title = element_blank()) +labs(x = "Gene", y = "Gene")
Ø安装和加载ggplot2包if (!requireNamespace("ggplot2", quietly = TRUE))install.packages("ggplot2")library(ggplot2)
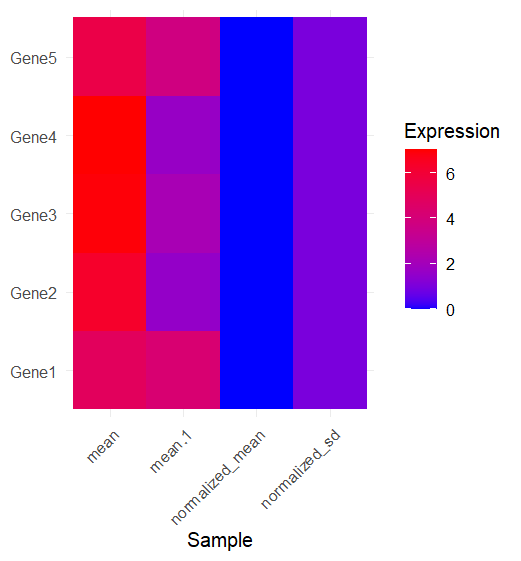
gene_info <- as.data.frame(gene_info)long_data <- tidyr::pivot_longer(gene_info, cols = -Gene, names_to = "Sample", values_to = "Expression")ggplot(long_data, aes(x = Sample, y = Gene, fill = Expression)) +geom_tile() +theme_minimal() +theme(axis.text.x = element_text(angle = 45, hjust = 1),axis.title.y = element_blank(),panel.border = element_blank()) +scale_fill_gradient(low = "blue", high = "red")
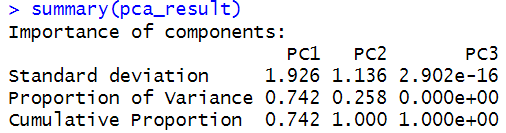
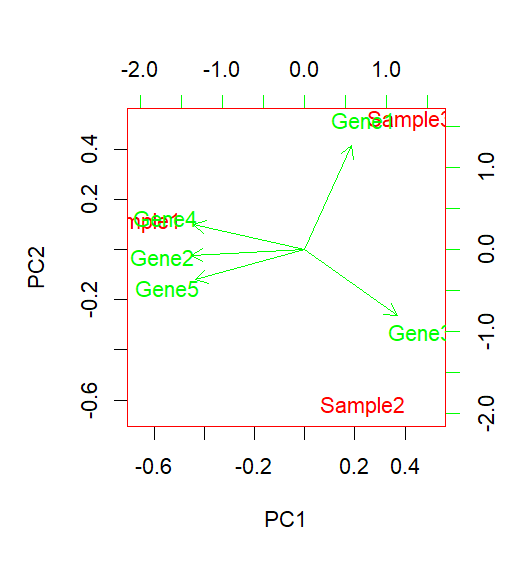
pca_result <- prcomp(t(expression_data), scale. = TRUE)summary(pca_result)biplot(pca_result, scale = TRUE, col = c("red", "green", "blue"))
if (!requireNamespace("tidyr", quietly = TRUE))install.packages("tidyr")library(tidyr)
long_gene_info <- pivot_longer(gene_info, cols = -Gene, names_to = "Statistic", values_to = "Expression")ggplot(long_gene_info, aes(x = Gene, y = Expression, fill = Gene)) +geom_boxplot() +theme_minimal() +theme(axis.text.x = element_text(angle = 45, hjust = 1)) +facet_wrap(~Gene, scales = "free_y") +labs(x = "gene", y = "Expression Value")
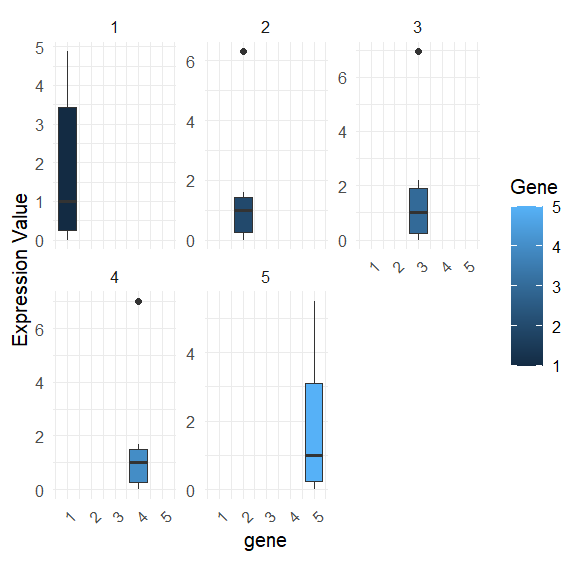
ggplot(long_gene_info, aes(x = Gene, y = Expression, color = Gene)) +geom_point(size = 2) +theme_minimal() +theme(axis.text.x = element_text(angle = 45, hjust = 1)) +facet_wrap(~Gene ,scales = "free_y") +labs(x = "Gene", y = "Expression Value")
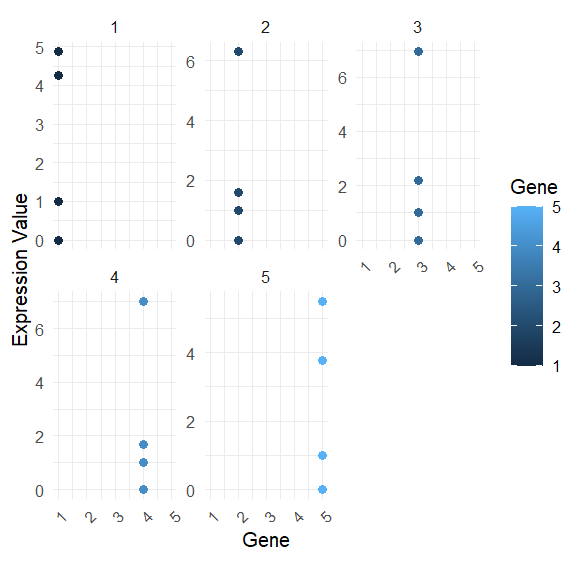
long_gene_info$Gene <- as.numeric(as.factor(long_gene_info$Gene))ggplot(long_gene_info, aes(x = Gene, y = Expression, color = Gene)) +geom_line() +geom_point() +theme_minimal() +labs(x = "Time Point", y = "Expression Value")
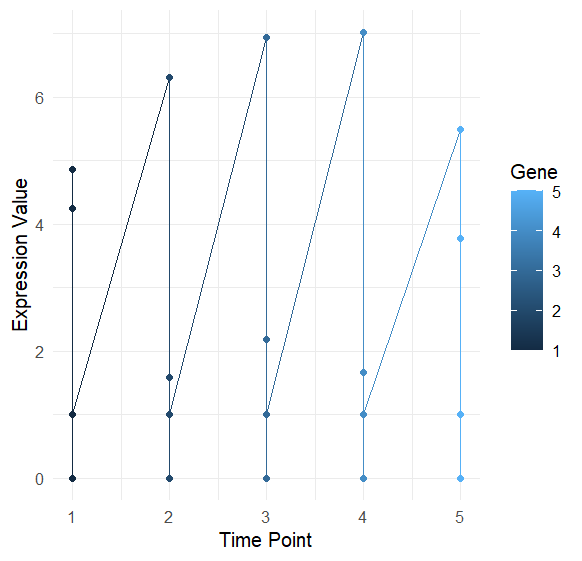
三、文章小结
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果

往期回顾
|
01 |
|
02 |
|
03 |
|
04 |