听说可以用R语言完成孟德尔随机化遗传相关,你来不来学?
点击蓝字
关注小图
简介:
该方法使用GWAS的概括性数据来估计一对表型的遗传相关。该方法基于以下的事实:GWAS检验中,对一个SNP效应量的估计通常也会包含与该SNP成LD的其他SNP的效应,也就是说一个与其他SNP成高LD的SNP,通常也会有更高的卡方检验量。而当我们把卡方检验量替换为,来自两个相关表型的GWAS的Z分数的乘积时,这个事实依然存在。所以基于此,我们可以将单表型的LD分数回归扩展成如下图所示的:
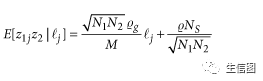
原理:
ldscr的目标是利用r中的LD评分回归,从GWAS汇总统计量中估算遗传度和跨性状遗传相关性。LD评分回归用于估算遗传度和遗传相关性的细节已经发表。该包改编了最初在 GenomicSEM中实现的代码和功能。
install.packages("devtools")devtools::install_github("mglev1n/ldscr")#最新版本的R无法安装成果,建议用R 4.1.1版的进行安装
数据介绍
ldsc_h2()可以用来估计遗传。
提供了示例GWAS数据sumstats_munged_example()。大家可以利用内置的LD参考数据进行演练,也可提供自己的数据。
数据文件储存在R包library中ldscr包的extdata文件夹里
#加载ldscrlibrary(ldscr)setwd("D:/R/R-4.1.1/library/ldscr")#根据自己的文件位置进行更改df <- sumstats_munged_example(example = "BMI")h2_res <- ldsc_h2(munged_sumstats = df, ancestry = "EUR")ldsc_rg可用于估计两个或多个性状之间的交叉性状遗传相关性rg_res <- ldsc_rg(munged_sumstats = list("APOB" = sumstats_munged_example(example = "APOB"),"LDL" = sumstats_munged_example(example = "LDL")),ancestry = "EUR")
rg_res#查看计算结果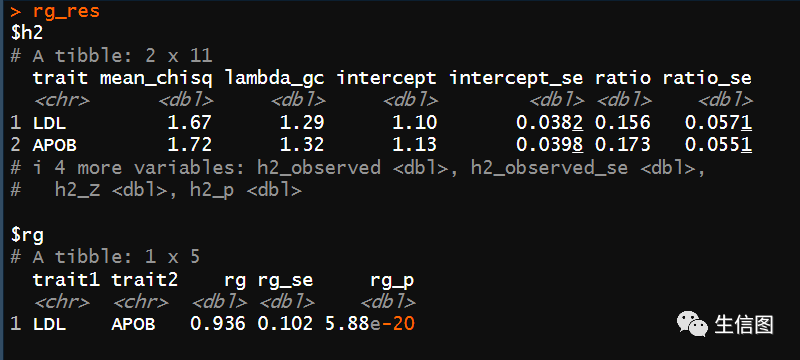
可见相关性:rg=0.936
显著性P值rg_pval=5.88e-20
大多数情况我们都需要根据自己分析的数据进行遗传相关性的计算,这时就需要下载并处理原始数据,那么原始数据如何进行处理呢
首先下载需要分析的数据
用R包gwasglue进行数据格式处理
这个R包充当可以读取或查询GWAS汇总数据的包和可以分析GWAS汇总数据的包之间的管道。以下是它在GWAS数据和分析的总体生态系统中的位置:
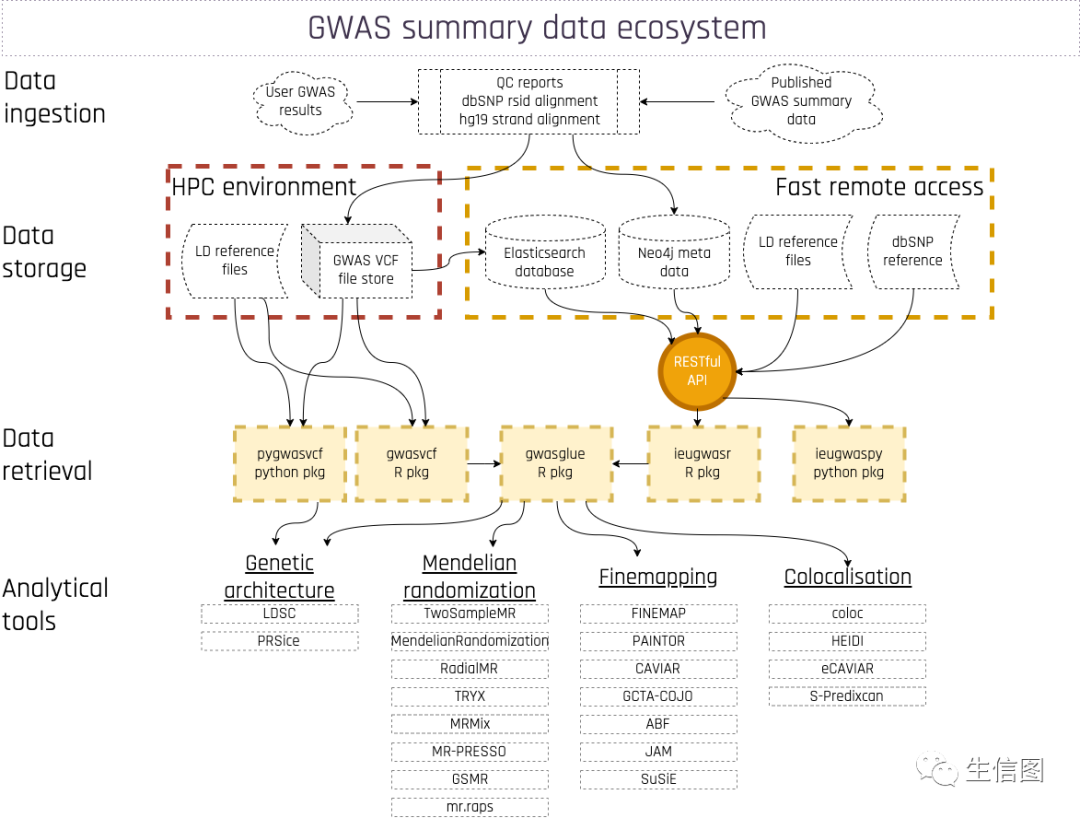
完整的GWAS汇总数据集现在非常丰富。在IEU GWAS数据库中有一个大型的数据库,包含了经过管理的、协调的和质量控制的数据集。他们可以通过API直接查询,或者通过ieugwasr R包,或ieugwaspy python包。但是,对于可以在HPC环境中使用的更快的查询,直接访问数据而不是通过云系统是有利的。
于是,开发了一种用于存储和协调GWAS汇总数据的格式,称为GWAS VCF格式,可以使用gwas2vcf创建。IEU GWAS数据库中的所有数据均可以这种格式下载。这个R包提供了快速便捷的查询和创建GWAS VCF格式的GWAS汇总数据的功能。
#安装处理VCF格式的R包if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("VariantAnnotation")library(VariantAnnotation)devtools::install_github("mrcieu/gwasglue")#加载library(gwasglue)library(TwoSampleMR)setwd("D:/R/R-4.1.1/library/ldscr")#根据自己文件情况设置路径dat<- readVcf("ukb-b-956.vcf.gz") #读入GWAS的summary结局/暴露数据的VCF格式文件dat <-gwasvcf_to_TwoSampleMR(dat)#转换为TwoSampleMR可以识别的格式
小图温馨提示:由于GWAS的summary数据文件大小再200-300MB左右,所以windows下的R可能读取失败,建议在linux下进行格式转换。
dat数据框第3,4,5,6,9,11列;这里分别对应的是”A2″,”A1″,”beta”,”se”,”N”,”SNP”
dat<- dat [, c(3:6,9,11)]colnames(dat)<-c("A2","A1","beta","se","N","SNP")#对数据文件重命名#最后将文件保存为txt.gz格式write.table(dat, gzfile("ukb-b-7178-sumstats-munged.txt.gz"),sep = "t", row.names = FALSE)
数据格式处理成功即可按照上述流程估算遗传度和遗传相关性
关于GWAS summary 数据遗传度和遗传相关性估算小图就介绍完毕了,希望大家能牢记各个文件的结构信息,这在后续的数据分析中非常重要。
欢迎使用:云生信生物信息学平台( http://www.biocloudservice.com/home.html),了解更多生信知识与技巧分析。
请期待小图的下一期孟德尔随机化知识分享。
欢迎使用:云生信平台 ( http://www.biocloudservice.com/home.html)

|
往期推荐 |
|
|
|
|
|
|
👇点击阅读原文进入网址