如果你还不会利用limma进行多组差异分析?不慌!跟着小果一文拿捏
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

今天小果想为大家分享的是基于limma包进行多组差异分析
limma包在进行GEO数据库mRNA芯片数据分析时,使用的频率非常非常高,强烈安利小伙伴学习奥!有GEO数据分析需求的小伙伴可以跟着小果一起来学习,接下来小果就开始今天的分享。
1. 如何利用limma进行多组差异分析?
在进行讲解如何进行分析之前,小果想为小伙伴简单讲解一下limma包的分析原理,该包是基于线性模型进行差异分析,如何使用该包进行转录组数据差异分析,可以参考官网http://bioconductor.org/packages/release/bioc/vignettes/limma/inst/doc/usersguide.pdf文档进行学习,小果在这里就不做详细介绍了。本推文只需要基因表达矩阵文件和样本分组文件,就可以批量完成多组差异分析,非常简单方便,值得小伙伴们学习奥!接下来跟着小果开始今天的实操学习吧!
2. 准备需要的R包
limma包的安装直接可以通过Bioconductor安装,安装非常简单,小果为大家也提供网址:http://bioconductor.org/packages/release/bioc/html/limma.html,如下:
#安装limma包BiocManager::install("limma")#加载limma包library(limma)
3. 数据准备
input_FPKM.txt
#基因表达矩阵文件,行名为Gene,列为样本名.
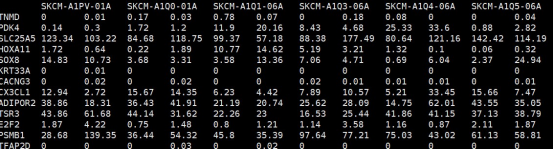
input_subtype.txt
##样本分组信息文件,第一列为样本名,第二列为分组信息

4. limma进行多组差异分析
# 创建需要配对比较的列表函数createList <- function(group=NULL) {tumorsam <- names(group)sampleList = list()treatsamList =list()treatnameList <- c()ctrlnameList <- c()#A-1: 类1 vs 其他sampleList[[1]] = tumorsamtreatsamList[[1]] = intersect(tumorsam, names(group[group=="immune"])) # 亚型名称需要根据情况修改treatnameList[1] <- "immune" # 该亚型的命名ctrlnameList[1] <- "Others" # 其他亚型的命名#A-2: 类2 vs 其他sampleList[[2]] = tumorsamtreatsamList[[2]] = intersect(tumorsam, names(group[group=="keratin"]))treatnameList[2] <- "keratin"ctrlnameList[2] <- "Others"#A-3: 类3 vs 其他sampleList[[3]] = tumorsamtreatsamList[[3]] = intersect(tumorsam, names(group[group=="MITF-low"]))treatnameList[3] <- "MITF-low"ctrlnameList[3] <- "Others"#如果有更多类,按以上规律继续写return(list(sampleList, treatsamList, treatnameList, ctrlnameList))}# 配对limma差异分析函数twoclasslimma <- function(res.path=NULL, expr=NULL, prefix=NULL, complist=NULL, overwt=FALSE) {sampleList <- complist[[1]]treatsamList <- complist[[2]]treatnameList <- complist[[3]]ctrlnameList <- complist[[4]]allsamples <- colnames(expr)options(warn=1)for (k in 1:length(sampleList)) { # 循环读取每一次比较的内容samples <- sampleList[[k]]treatsam <- treatsamList[[k]]treatname <- treatnameList[k]ctrlname <- ctrlnameList[k]compname <- paste(treatname, "_vs_", ctrlname, sep="") # 生成最终文件名tmp = rep("others", times=length(allsamples))names(tmp) <- allsamplestmp[samples]="control"tmp[treatsam]="treatment"outfile <- file.path( res.path, paste(prefix, "_limma_test_result.", compname, ".txt", sep="") )if (file.exists(outfile) & (overwt==FALSE)) { # 因为差异表达分析较慢,因此如果文件存在,在不覆盖的情况下(overwt=F)不再次计算差异表达cat(k, ":", compname, "exists and skipped;n")next}pd <- data.frame(Samples=names(tmp),Group=as.character(tmp),stringsAsFactors = F)design <-model.matrix(~ -1 + factor(pd$Group, levels=c("treatment","control"))) # 设计矩阵仅包含亚型信息,若有批次效应请修改colnames(design) <- c("treatment","control")# 对数转换gset <- log2(expr + 1)# 差异表达过程fit <- lmFit(gset, design=design);contrastsMatrix <- makeContrasts(treatment - control, levels=c("treatment", "control"))fit2 <- contrasts.fit(fit, contrasts=contrastsMatrix)fit2 <- eBayes(fit2, 0.01)tT <- topTable(fit2, adjust="fdr", sort.by="B", number=100000)tT <- subset(tT, select=c("logFC","t","B","P.Value","adj.P.Val"))colnames(tT) <- c("log2FC","t","B","PValue","FDR")tT <- tT[order(tT$FDR),]write.table(tT, file=outfile, row.names=T, col.names=NA, sep="t", quote=F)cat(k, ",")}options(warn=0)}# 读取表达矩阵expr <- read.table("input_FPKM.txt",sep = "t",header = T,check.names = F,stringsAsFactors = F,row.names = 1)# 读取分组信息subt <- read.table("input_subtype.txt", sep = "t", check.names = F, stringsAsFactors = F, header = T, row.names = 1)#分组名称n.sub.label <- unique(subt$TCGA_Subtype)#分组个数n.sub <- length(table(subt$TCGA_Subtype))### 创建配对比较的列表信息#获得分组名group <- subt$TCGA_Subtype#通过样本名来命名names(group) <- rownames(subt)#利用createList函数创建需要比较的列表信息complist <- createList(group=group)# 利用twoclasslimma函数进行分组差异分析,所有配对差异表达结果都会输出在res.path路径下。twoclasslimma(res.path = ".",expr = expr[,intersect(colnames(expr),rownames(subt))],prefix = "SKCM",complist = complist,overwt = F)
5. 结果文件
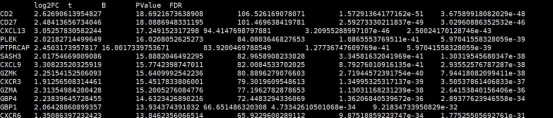
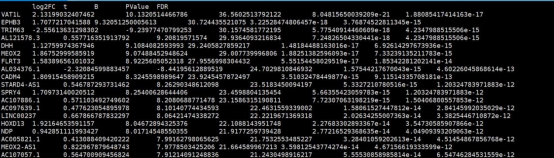
1. SKCM_limma_test_result.immune_vs_Others.txt
该结果文件为计算的immune组与其他分组的差异分析结果文件,行名为基因名,第一列为log2FC,第四列为Pvalue值,第五列为FDR(矫正后的P值)
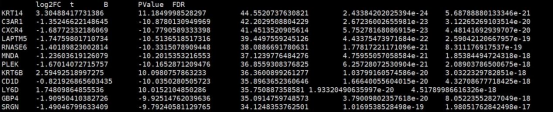
2. SKCM_limma_test_result.keratin_vs_Others.txt
该结果文件为计算的keratin组与其他分组的差异分析结果文件,行名为基因名,第一列为log2FC,第四列为Pvalue值,第五列为FDR(矫正后的P值)
3. SKCM_limma_test_result.MITF-low_vs_Others.txt
该结果文件为计算的MITF-low组与其他分组的差异分析结果文件,行名为基因名,第一列为log2FC,第四列为Pvalue值,第五列为FDR(矫正后的P值).
今天小果的分享就到这里啦!如果小伙伴有其他数据分析需求,可以尝试使用本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:http://www.biocloudservice.com/home.html,包括了用edgeR实现多组差异分析(http://www.biocloudservice.com/289/289.php),DEseq2实现多组差异分析(http://www.biocloudservice.com/287/287.php)等小工具,欢迎大家和小果一起讨论学习哈!!!!
往期推荐

点击“阅读原文”立刻拥有
↓↓↓