手把手教你miRNA生存相关分析绘图
收录于话题
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

微小RNA的表达数据与患者的生存数据进行分析,以寻找与患者生存相关的miRNA。生存分析和绘制生存曲线,找出与患者生存相关的miRNA。这对于了解miRNA在癌症等疾病预后中的作用以及寻找潜在的生物标志物具有重要意义。生存分析是一种常用的统计分析方法,可用于评估不同基因或生物分子在患者预后中的潜在影响。通过该脚本,研究人员可以快速而全面地对大规模miRNA表达数据进行生存相关分析,从而辅助癌症等疾病的研究和诊断。
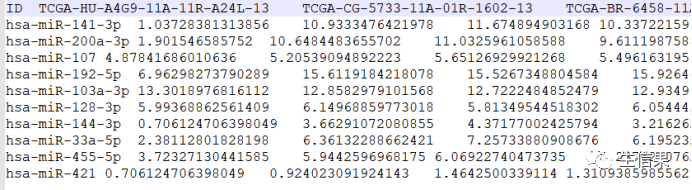
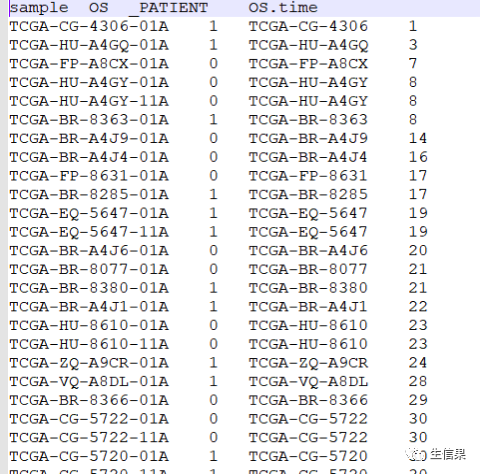
代码中对选定的相关miRNA的表达量进行生存分析。下面是要研究的多个mirna的样本表达量,以及下载的STAD癌症的生存时间和状态。
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("limma")
检查是否已安装”BiocManager”包,如果没有安装,则通过install.packages函数安装它。
“BiocManager”包用于管理和安装生物信息学相关的其他包。
install.packages('survival')install.packages("survminer")
安装”survminer”包,它是一个用于生存数据可视化和统计分析的包。
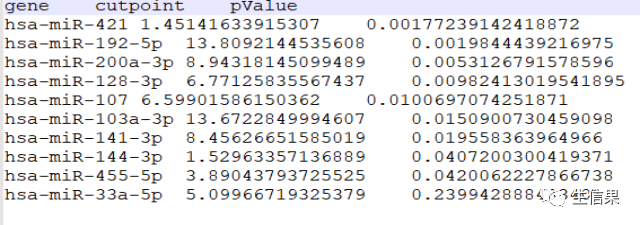
library(limma)library(survival)library(survminer)expFile="cor.MirnaExp.txt"cliFile="TCGA-STAD.survival.tsv"data=read.table(expFile, header=T, sep="t", check.names=F, row.names=1)group=sapply(strsplit(colnames(data),"\-"), "[", 4)group=sapply(strsplit(group,""), "[", 1)group=gsub("2", "1", group)data=t(data[,group==0,drop=F])rownames(data)=gsub("(.*?)\-(.*?)\-(.*?)\-(.*?)\-.*", "\1\-\2\-\3-\4", rownames(data))cli=read.table(cliFile, header=T, sep="t", check.names=F, row.names=1)cli=cli[,c(3,1)]cli=na.omit(cli)colnames(cli)=c("futime","fustat")cli$futime=cli$futime/365check.names=F表示不检查列名的合法性。row.names=1表示使用第一列作为行名。sameSample=intersect(row.names(data), row.names(cli))data=data[sameSample,,drop=F]cli=cli[sameSample,,drop=F]rt=cbind(cli, data)outTab=data.frame()for(gene in colnames(rt)[3:ncol(rt)]){if(sd(rt[,gene])<0.1){next}data=rt[,c("futime", "fustat", gene)]colnames(data)=c("futime", "fustat", "gene")res.cut=surv_cutpoint(data, time = "futime", event = "fustat", variables =c("gene"))res.cat=surv_categorize(res.cut)fit=survfit(Surv(futime, fustat) ~gene, data = res.cat)diff=survdiff(Surv(futime, fustat) ~gene, data =res.cat)pValue=1-pchisq(diff$chisq, df=1)outVector=cbind(gene, res.cut$cutpoint[1], pValue)outTab=rbind(outTab,outVector)if(pValue<0.001){pValue="p<0.001"}else{pValue=paste0("p=",sprintf("%.03f",pValue))}surPlot=ggsurvplot(fit,data=res.cat,pval=pValue,pval.size=6,legend.title=gene,legend.labs=c("high","low"),xlab="Time(years)",ylab="Overall survival",palette=c("red", "blue"),break.time.by=1,conf.int=T,risk.table=F,risk.table.title="",risk.table.height=.25)pdf(file=paste0("sur.",gene,".pdf"),onefile = FALSE,width = 5.5,height =5)print(surPlot)dev.off()}outTab=outTab[order(as.numeric(as.vector(outTab[,"pValue"]))),]write.table(outTab, file="survival.txt", sep="t", row.names=F, quote=F)
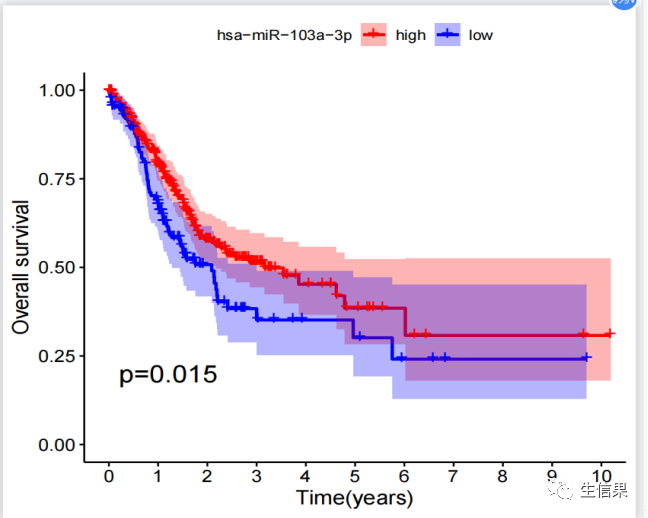
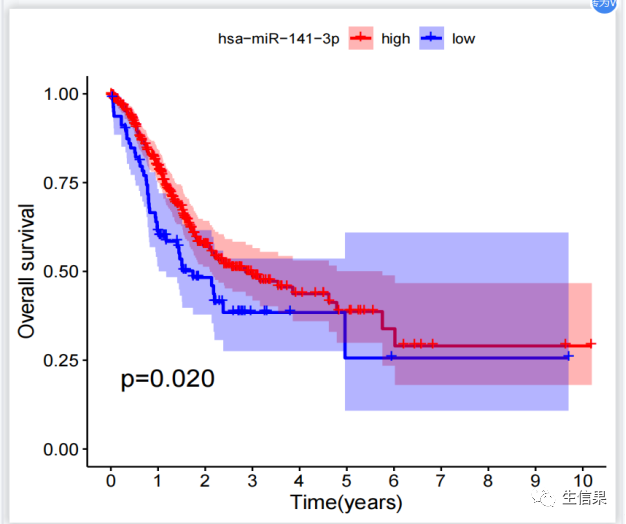
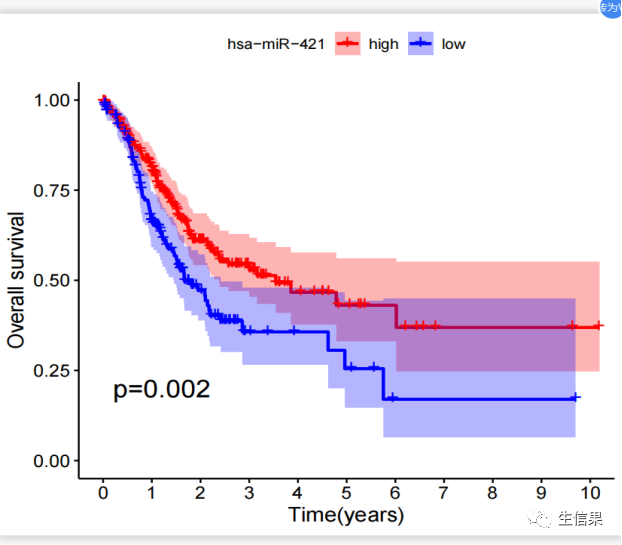
下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:http://www.biocloudservice.com/home.html。
云生信平台链接:http://www.biocloudservice.com/home.html。
其他相关分析内容,例如预测肿瘤样本药物敏感性分析(http://www.biocloudservice.com/712/712.php),预测某样本亚型对免疫治疗的反应(http://www.biocloudservice.com/292/292.php),单样本富集算法分析免疫浸润丰度(http://www.biocloudservice.com/106/106.php),计算64种免疫细胞相对含量(http://www.biocloudservice.com/107/107.php)等都可以用本公司新开发的零代码云平台生信分析小工具,一键完成该分析奥,感兴趣的小伙伴欢迎来尝试奥,网址:http://www.biocloudservice.com/home.html。今天小果的分享就到这里,下期在见奥。
小果友情推荐
好用又免费的工具安利



点击“阅读原文”立刻拥有
↓↓↓