scMerge2魔法指南:小果带你领略单细胞数据融合的奇妙之旅!
 前言
前言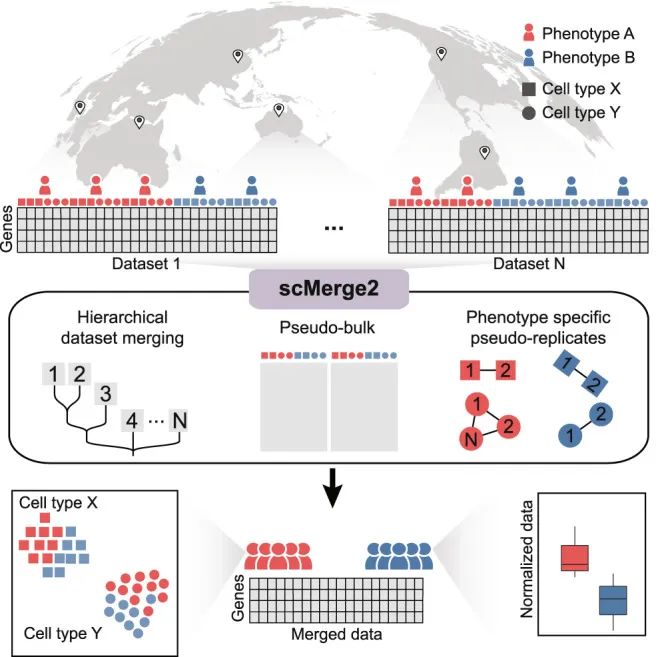
## 从 Bioconductor 安装 scMerge,需要 R 3.6.0 或以上版本BiocManager::install("scMerge")##您也可以尝试安装 ScMerge 的 Bioconductor 开发版BiocManager::install("scMerge", version = "devel")
suppressPackageStartupMessages({library(SingleCellExperiment)library(scMerge)library(scater)})
## 小鼠 ESC 数据子集data("example_sce", package = "scMerge")data("segList_ensemblGeneID", package = "scMerge")
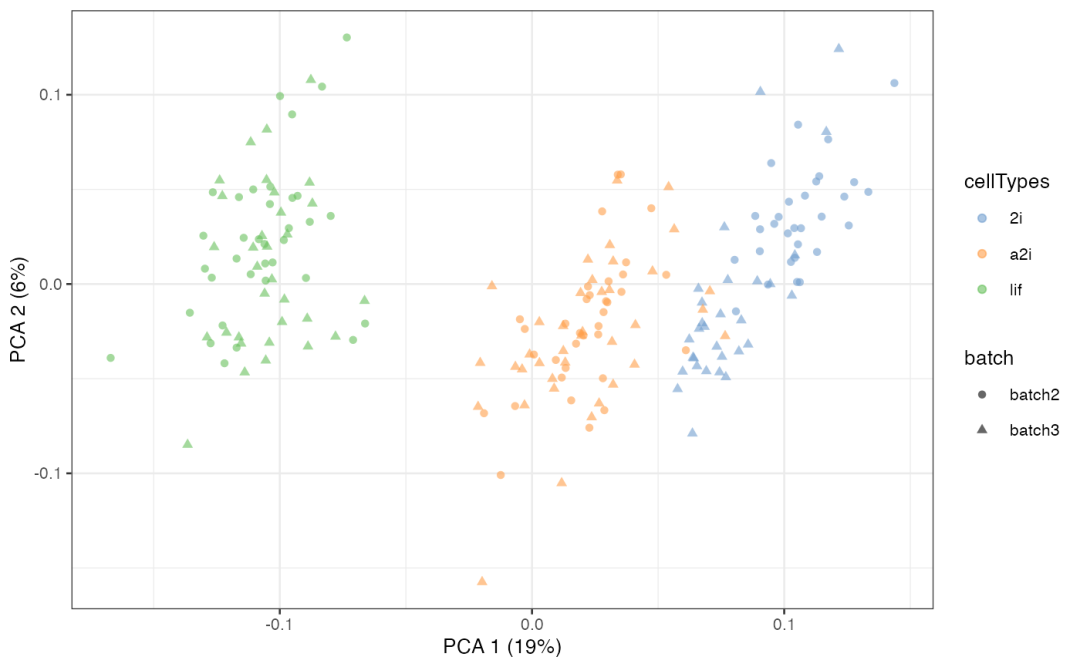
example_sce = runPCA(example_sce, exprs_values = "logcounts")scater::plotPCA(example_sce,colour_by = "cellTypes",shape_by = "batch")

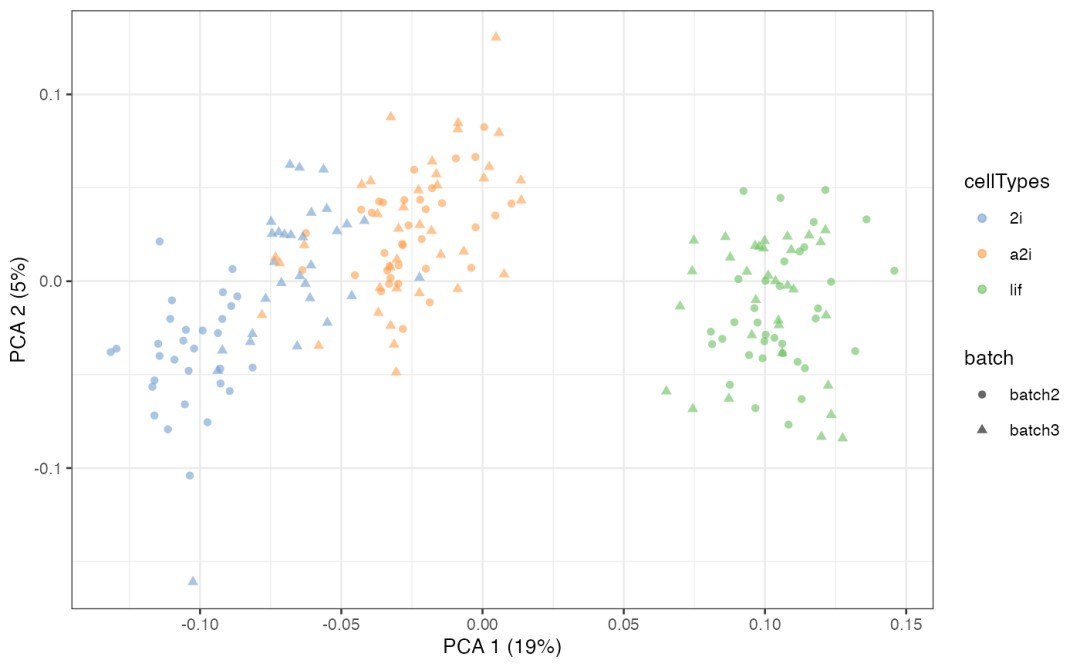
scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),batch = example_sce$batch,ctl = segList_ensemblGeneID$mouse$mouse_scSEG,verbose = FALSE)assay(example_sce, "scMerge2") <- scMerge2_res$newYset.seed(2022)example_sce <- scater::runPCA(example_sce, exprs_values = 'scMerge2')scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')
scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),batch = example_sce$batch,cellTypes = example_sce$cellTypes,ctl = segList_ensemblGeneID$mouse$mouse_scSEG,verbose = FALSE)assay(example_sce, "scMerge2") <- scMerge2_res$newYexample_sce = scater::runPCA(example_sce, exprs_values = 'scMerge2')scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')

scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),batch = example_sce$batch,ctl = segList_ensemblGeneID$mouse$mouse_scSEG,k_pseudoBulk = 50,verbose = FALSE)assay(example_sce, "scMerge2") <- scMerge2_res$newYset.seed(2022)example_sce <- scater::runPCA(example_sce, exprs_values = 'scMerge2')scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')
 在处理大量数据时,我们可以每次获取较小单元格子集的调整后矩阵。这可以通过在in函数中设置,函数将不返回调整后的整个矩阵,而是输出估计值。然后,为了使用估计值得到调整后的矩阵,我们首先需要对对数矩阵进行余弦归一化处理,然后计算余弦归一化矩阵的行向(基因向)平均值(这是因为默认情况下,在步骤之前会对对数归一化矩阵进行余弦归一化处理)。然后,每次我们都可以用它来调整单元格子集的矩阵。
在处理大量数据时,我们可以每次获取较小单元格子集的调整后矩阵。这可以通过在in函数中设置,函数将不返回调整后的整个矩阵,而是输出估计值。然后,为了使用估计值得到调整后的矩阵,我们首先需要对对数矩阵进行余弦归一化处理,然后计算余弦归一化矩阵的行向(基因向)平均值(这是因为默认情况下,在步骤之前会对对数归一化矩阵进行余弦归一化处理)。然后,每次我们都可以用它来调整单元格子集的矩阵。scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),batch = example_sce$batch,ctl = segList_ensemblGeneID$mouse$mouse_scSEG,verbose = FALSE,return_matrix = FALSE)cosineNorm_mat <- batchelor::cosineNorm(logcounts(example_sce))adjusted_means <- DelayedMatrixStats::rowMeans2(cosineNorm_mat)newY <- list()for (i in levels(example_sce$batch)) {newY[[i]] <- getAdjustedMat(cosineNorm_mat[, example_sce$batch == i],scMerge2_res$fullalpha,ctl = segList_ensemblGeneID$mouse$mouse_scSEG,ruvK = 20,adjusted_means = adjusted_means)}newY <- do.call(cbind, newY)assay(example_sce, "scMerge2") <- newY[, colnames(example_sce)]set.seed(2022)example_sce <- scater::runPCA(example_sce, exprs_values = 'scMerge2')scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')
# 创建一个假样本信息example_sce$sample <- rep(c(1:4), each = 50)table(example_sce$sample, example_sce$batch)
# 构建分层索引列表h_idx_list <- list(level1 = split(seq_len(ncol(example_sce)), example_sce$batch),level2 = list(seq_len(ncol(example_sce))))
h_idx_list$level1
h_idx_list$level2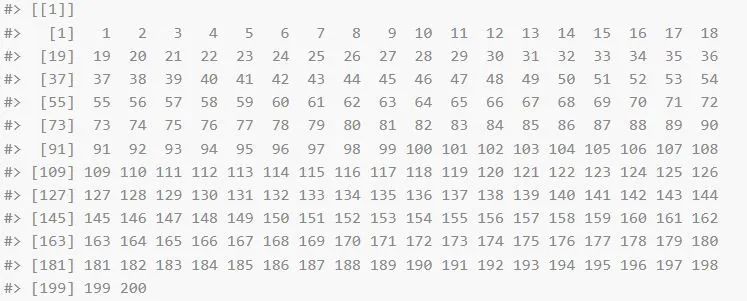
# 构建批次信息列表batch_list <- list(level1 = split(example_sce$sample, example_sce$batch),level2 = list(example_sce$batch))
batch_list$level1
batch_listh_idx_listscMerge2hruvK_listruvKruvK_list = c(2, 5)scMerge2_res <- scMerge2h(exprsMat = logcounts(example_sce),batch_list = batch_list,h_idx_list = h_idx_list,ctl = segList_ensemblGeneID$mouse$mouse_scSEG,ruvK_list = c(2, 5),verbose = FALSE)
length(scMerge2_res)lapply(scMerge2_res, dim)
assay(example_sce, "scMerge2") <- scMerge2_res[[length(h_idx_list)]]set.seed(2022)example_sce <- scater::runPCA(example_sce, exprs_values = 'scMerge2')scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')

h_idx_list2 <- h_idx_listbatch_list2 <- batch_listh_idx_list2$level1$batch2 <- NULLbatch_list2$level1$batch2 <- NULLprint(h_idx_list2)

print(batch_list2)
scMerge2_res <- scMerge2h(exprsMat = logcounts(example_sce),batch_list = batch_list2,h_idx_list = h_idx_list2,ctl = segList_ensemblGeneID$mouse$mouse_scSEG,ruvK_list = c(2, 5),verbose = FALSE)assay(example_sce, "scMerge2") <- scMerge2_res[[length(h_idx_list)]]set.seed(2022)example_sce <- scater::runPCA(example_sce, exprs_values = 'scMerge2')scater::plotPCA(example_sce, colour_by = 'cellTypes', shape = 'batch')



往期推荐