震惊!这个快速找变异的方法你还不知道?
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

各位生信狗们好呀,这里是攒钱买米诺地尔的小果子,今天咱来唠唠一个神奇的黑科技:BSA分析(Bulked Segregant Analysis)。
咱就是说,在咱生物学领域,基因突变一直是最关键的内容之一。为了找到影响生物表型变异的基因,咱从QTL做到GWAS(都给我心疼小果三分钟,小果可是做林木的),工作繁琐而费时,但是,BSA的横空出世,为寻找基因突变提供了一种更快、更便宜的方法。
BSA简介:
首先BSA需要构建遗传群体(F2、BC、RIL),它的原理是将群体中的极端样本进行测序,从而找出极端样本之间的遗传差异。该分析对穷逼课题组极其友好(回想当初小果的课题组,呆过的人才知道个中滋味),仅需要对亲本进行重测序,子代的极端样本混合成两个混池(为什么是两个?废话,极端肯定有极大极小两个啊),将混池按照样本数量和基因组大小进行测序(怎么样,是不是很便宜,GWAS测不起的苦有谁懂啊)。
BSA的原理:
SNP-index作为主流的BSA定位的算法,在2013年由Takagi提出(热乎的新分析哟)。其原理为,子代分离群体中极端性状的样本构建混池后以亲本为参考基因组进行SNP calling,然后分析两个混池等位基因频率。与参考基因组不同的基因型的比例,就是为该位点的SNP-index。从下图可以看到,两个位点的SNP-index分别为0.4和1。SNP-index在1和-1处的峰即为与性状相关的SNP。

实际情况当然不可能是理想状态,比如林木构建分析群体困难(小果我哭死),亲本数据缺失等。这时候就需要万能的欧氏距离出马了(how old are you)。
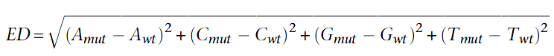
欧氏距离计算混池间的等位基因频率,原理与SNP-index法类似,在实际分析中,我们会对一个滑窗内所有位点的ED值进行拟合,消除抽样偏差产生的假阳性。再去ED值的平方,放大ED值的差异,使定位区间更加明显。

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓