单细胞测序流程(三):单细胞计数矩阵的生成
上一期小果跟大家唠了单细胞测序的建库流程,那么这一回,咱们来唠唠单细胞计数矩阵的生成。
上回书说到,在建库时,单细胞测序通过barcode区分来源不同细胞的RNA序列,但是在同一个细胞中,同一基因可能会产生多次转录,而PCR步骤也会产生重复读取。为了区分reads是来源于生物扩增还是PCR扩增,scRNA-seq采用在PCR扩增前在cDNA序列上引入UMI标记,确定单个cDNA扩增过程中产生的拷贝数。因此scRNA-seq的数据处理有以下几种可能
相同转录本但UMI不同的reads来源于同一细胞不同的分子,属于生物学重复,每个reads都应当被计算。
具有相同UMI的reads来源于同一分子的PCR重复,应当被即为单个reads。
如图所示,ATCB应当被记为单词reads,而ARL1应当分别计数。
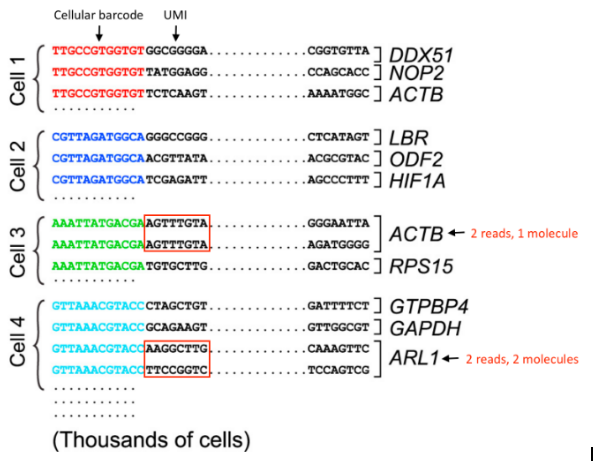
了解UMI的原理,有利于我们了解scRNA-seq如何在细胞水平上进行量化。
在了解了UMI和barcode是如何区分不同细胞和不同分子后,接下来咱们来了解以下scRNA-seq的工作流程。
工作流程步骤为:
计数矩阵的生成:formating reads, demultiplexing samples, mapping and quantification
原始计数矩阵的质控:过滤劣质细胞
聚类:基于转录活性的相似性对细胞进行聚类(细胞类型 类似于 不同的clusters)
marker鉴定和簇注释:识别每个簇的marker并注释已知的细胞类型簇。
工作流程如下图所示:
:单细胞计数矩阵的生成/2_ZWrfEQ5iahxXG67lX1ToKpkakbkNFg.png)
当然,无论进行什么分析,基于不同条件的单个样本得出的关于总体的结论都是不可信的。若想得到可靠的结论,仍然需要生物重复!也就是说,如果您想得出与总体相对应的结论,请做生物学重复。
在完成了测序后,原始测序数据一般输出为BCL或FASTQ格式。如果reads是BCL格式,可以使用cellranger的mkfastq工具将BCL转换为FASTQ格式。
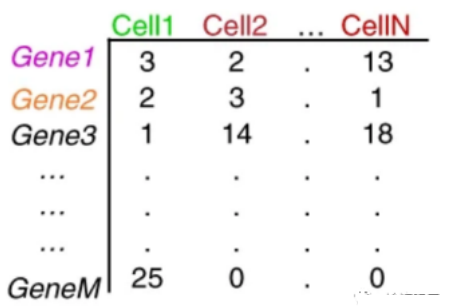
在得到FASTQ文件后,使用cell ranger软件进行数据的质控和定量,可以通过下图的细胞基因计数矩阵探索和过滤数据,输出的csv文件就可以使用R的seurat包进行下游分析啦。
下一回就到了激动人心的实操环节啦,不要走开,后续更精彩哟。
小果今天的分享就到这里,欢迎大家和小果一起讨论学习哦!我们下期再见~
小果友情推荐
好用又免费的工具安利


:单细胞计数矩阵的生成/6_IHSYtLyxwVWxiaf2v2rQdOnoz05G6A.jpg)