小果带你玩转单细胞RNA-seq数据:如何巧用Clustree包划分聚类亚群!
 小伙伴们大家好,很开心又能和大家分享分享小果在分析单细胞数据过程中的经验,今天来给大家说说clustree,不知道大家还记不记得小果之前介绍的Seurat包,这个包算是单细胞RNA-seq分析的始祖,它包括了数据加载和预处理,数据质量控制,归一化和标准化,降维,聚类,细胞注释等等多个部分,这就使得Seurat在当前单细胞数据分析中至关重要,因为它实现了端到端的一体化分析流程,但是我们可以从官网上看到,它在部分步骤上是没有优势的,比如批次效应的去除,细胞亚群的注释,以及我们今天想说的如何划分聚类亚群,因此现在也出现了很多与单细胞有关的包,这些包是Seurat的补充,大家分析的时候也要具体问题具体分析,不能全篇套Seurat, 还应该紧跟前沿,及时更新自己的分析流程工具~
小伙伴们大家好,很开心又能和大家分享分享小果在分析单细胞数据过程中的经验,今天来给大家说说clustree,不知道大家还记不记得小果之前介绍的Seurat包,这个包算是单细胞RNA-seq分析的始祖,它包括了数据加载和预处理,数据质量控制,归一化和标准化,降维,聚类,细胞注释等等多个部分,这就使得Seurat在当前单细胞数据分析中至关重要,因为它实现了端到端的一体化分析流程,但是我们可以从官网上看到,它在部分步骤上是没有优势的,比如批次效应的去除,细胞亚群的注释,以及我们今天想说的如何划分聚类亚群,因此现在也出现了很多与单细胞有关的包,这些包是Seurat的补充,大家分析的时候也要具体问题具体分析,不能全篇套Seurat, 还应该紧跟前沿,及时更新自己的分析流程工具~
既然我们今天要说的聚类亚群,那必然还是要从头说起,这次使用的数据是来自10XGenomics官网上的小鼠数据
(https://www.10xgenomics.com/resources/datasets/1k-mouse-kidney-nuclei-isolated-with-chromium-nuclei-isolation-kit-3-1-standard)
大家先跟着小果下载好我们就开始啦!
library(dplyr)library(Seurat)library(patchwork)library(ggplot2)pbmc.data<-Read10X(data.dir = "./ filtered_feature_bc_matrix")pbmc<-CreateSeuratObject(counts = pbmc.data,project = "pbmc3k",min.cells = 3,min.features = 200)pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")pbmc<-subset(pbmc,subset=nFeature_RNA>200 & nFeature_RNA<2500 & percent.mt<5)pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)all.genes <- rownames(pbmc)pbmc<-FindVariableFeatures(pbmc,selection.method = "vst",nfeatures =10)pbmc <- ScaleData(pbmc, features = all.genes)pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))ElbowPlot(pbmc)pbmc <- FindNeighbors(pbmc, dims = 1:5)pbmc <- FindClusters(pbmc, resolution = 0.1)pbmc<-RunUMAP(pbmc,dims = 1:5)DimPlot(pbmc,reduction = "umap")
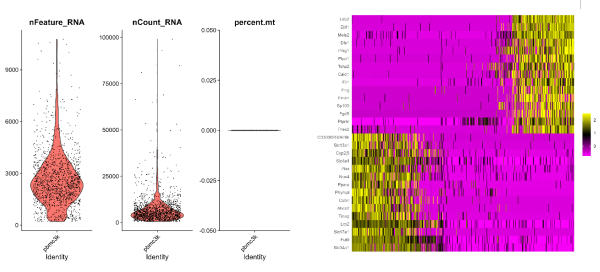
(顺便小果给大家附上两张Seurat结果图)
首先是导入数据,然后创建一个Seurat对象,接下来对数据进行质控(选择基因,细胞,标准化..),之后就要开始正式的分析了,先归一化,再PCA降维,然后就要开始聚类啦,那为什么要先降维再聚类呢,那是因为降维可以让数据降维到x轴和y轴上两个维度,这样也更方便聚类,因此RunPCA后就是FindNeighbors,参数如果选择dim为10,就是在10个PCA维度上找邻居,找到邻居根据一些图的算法才能聚类,说到这里,终于到了我们今天要说的函数了!FindClusters(pbmc,resolution=0.1),这是一个聚类分亚群的关键函数,resolution是本次内容的重点!
相信大家都知道,Resolution越高,对应的cluster可能就会越多,亚群分的越多就会带来信息过多,没有意义。而如果Resolution越小,则cluster越少,会丢失很多信息,后续的亚群注释也难以进行,由此可见,resolution的设置在整个实验过程中是十分重要的~
接下来小果给大家展示一下究竟是不是随着resolution的增高,cluster也会变多,在代码中我们将resolution从0.1到0.9进行循坏,通过dimplot来绘制降维的cluster图。
resolution_values <- seq(0.1, 0.9, by = 0.1)plots <- list() # 创建一个列表来存储图for (resolution in resolution_values) {# 执行聚类pbmc <- FindClusters(pbmc, resolution = resolution)# 绘制UMAP图plot <- DimPlot(pbmc, reduction = "umap")# 添加标题plot <- plot + ggtitle(paste("Resolution =", resolution))plots[[length(plots) + 1]] <- plot}# 使用cowplot包排列图final_plot <- plot_grid(plotlist = plots, ncol = 3)# 显示最终的排列图final_plot

 在本次的数据中虽然不是很明显,但是也可以看到差异,一开始是3个cluster,而之后变为了4个cluster,这也证实了小果之前说的是对的!resolution的选择十分关键,而我们该如何去做一个最恰当的选择呢?
在本次的数据中虽然不是很明显,但是也可以看到差异,一开始是3个cluster,而之后变为了4个cluster,这也证实了小果之前说的是对的!resolution的选择十分关键,而我们该如何去做一个最恰当的选择呢?
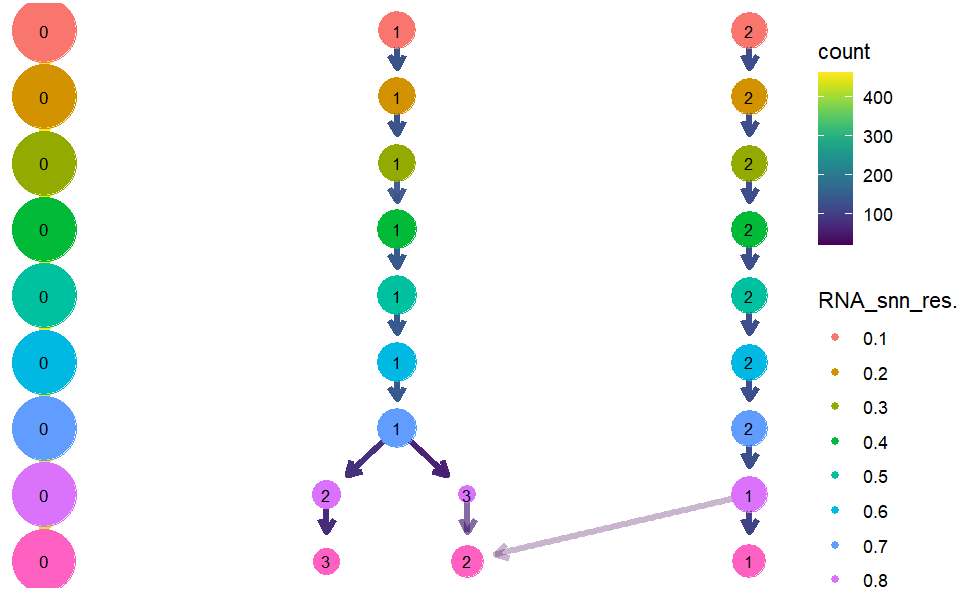
接下来给大家看看如何使用clustree包来判断应该到底取几个cluster.运行的,代码很简单:clustree(pbmc)就可以啦,如果还想加入一些别的参数来美化这个图,需要先看看Metadata的colnames:

clustree(pbmc@meta.data,prefix = "RNA_snn_res.")prefix可以选择metadata中的前缀要对比的内容,比如这里是所有的分辨率。当然,还有一些其他参数,大家可以自己查一查,包括颜色等等,最后可以美化的很好看哦
最后结果如下图所示,那我们看着这个图该怎么选择哪个分辨率呢,首先可以看到,图上有很多圈,每个圈的大小代表分到这个cluster中有多少细胞,不同的cluster在圈上都有标注,而每个圈的颜色则是对应的分辨率,比如在这里总共从0.1到0.8有8种分辨率。箭头则代表进一步细分,当我们选择的时候,更倾向于选择进一步细分的分辨率,比如红色框标注的那样,但是蓝色框这种一般就不可取,不太可靠。
为什么不太可靠呢?首先大家应该都知道,细胞之所以划分为不同的簇,是因为细胞群间的异质性大于细胞群内的异质性,但是如果蓝色框这样标注,意味着细胞群内的异质性大于群间的异质性,这明显与我们的假设不符。
因此我们可以通过这种方式进行细分,判断分辨率多少才是合适的,总而言之我们期望看到的是,亚群不断的细分,但是并不会跨细胞亚群进行划分。但是还是要去看对应的生物学功能,不断调整分辨率,再通过聚类树进行判断。
小果来总结一下!本数据中我们更希望的是resolution为0.7,因为再细分的话可能会出现细胞交叉的情况,没有意义,其实如何选择resolution并不会对结果产生太大的影响,只要选择合适,分析结果粗略可以看出就可以。


那怎么才能让聚类结果更有生物学意义呢?小果在这里引用一篇有关聚类树的论文进行说明,下面这张图分别以四种细胞作为marker,以颜色来表示这些marker的表达量,那怎么让表达量来覆盖颜色?这个就要看之前提到的参数了,在clustrr中有一个expr的属性,可以设置它的值来改变每个cluster(每个圈)的颜色,当已知意义的marker被聚类时,clustree中显示的最有颜色区分度的行就是我们想要的分辨率,大家可以试试怎么复现这篇文章的这幅图,论文名字也放这里啦:Clustering trees: a visualization for evaluating clusterings at multiple resolutions。

有帮助就给小果点个赞和在看,小果会继续努力的!


往期推荐