基因数据存在污染?不要怕!三分钟教会你使用decontam包,让你的数据干净无比!!

公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240314
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("decontam") # 在BiocManager环境下安装decontam查看是否安装成功packageVersion("decontam") # 查看decontam版本

library(phyloseq); packageVersion("phyloseq") # 载入phyloseq包并查看phyloseq包版本。library(ggplot2); packageVersion("ggplot2") # 载入ggplot2包并查看ggplot2包版本。library(decontam); packageVersion("decontam") # 载入decontam包并查看decontam包版本。

ps <- readRDS(system.file("extdata", "MUClite.rds", package="decontam")) # 提取MUClite.rds文件数据到ps变量ps # 显示ps数据信息

head(sample_data(ps)) # 查看原始样本数据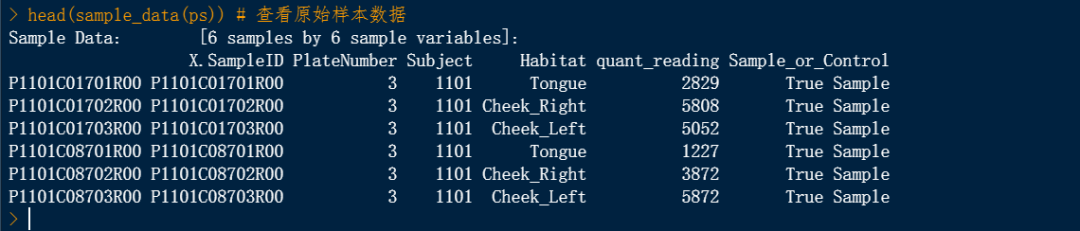
contamdf.freq <- isContaminant(ps, method="frequency", conc="quant_reading") # 使用frequency方法识别污染序列head(contamdf.freq) # 显示结果显示如下图所示:

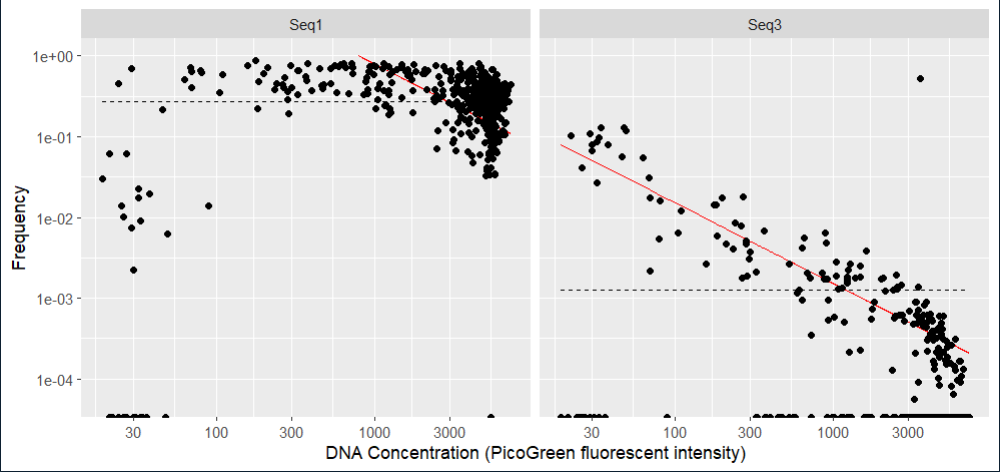
set.seed(100) # 设置参数plot_frequency(ps, taxa_names(ps)[sample(which(contamdf.freq$contaminant),3)], conc="quant_reading") + xlab("DNA Concentration (PicoGreen fluorescent intensity)") # 做出被判定为污染物的ASV的frequences 分布图

ps.noncontam <- prune_taxa(!contamdf.freq$contaminant, ps) # 去除污染物,赋值给新的变量。ps.noncontam # 展示去除结果

小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |