难道你不想一秒钟看透基因组的秘密吗?maftools包,让研究基因组突变不再是难题!


公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240423
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

maftools包的介绍
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager ")BiocManager::install("maftools") # 在BiocManager环境下安装maftools查看是否安装成功packageVersion("maftools") # 查看maftools版本

library(maftools) # 载入maftools包laml.maf = system.file('extdata', 'tcga_laml.maf.gz', package = 'maftools') # 载入“tcga_laml.maf.gz”数据laml.clin = system.file('extdata', 'tcga_laml_annot.tsv', package = 'maftools') # 载入“tcga_laml_annot.tsv”数据laml = read.maf(maf = laml.maf, clinicalData = laml.clin) # 将两个数据组合到变量laml中
class(laml) # 查看laml数据类型
laml # 显示laml整体信息
getSampleSummary(laml) # 显示样本突变信息
getGeneSummary(laml) # 显示基因突变信息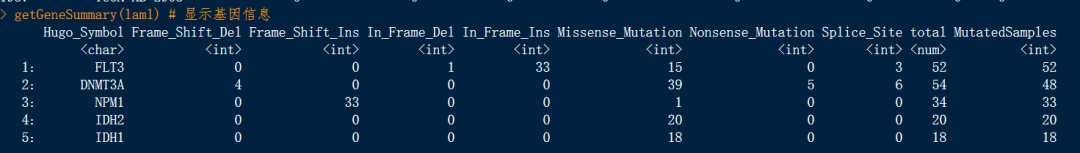
plotmafSummary(maf = laml, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE, titvRaw = FALSE) # 画出堆叠条形图和箱线图
oncoplot(maf = laml, top = 10) # 对排名前十的基因绘制waterfall图 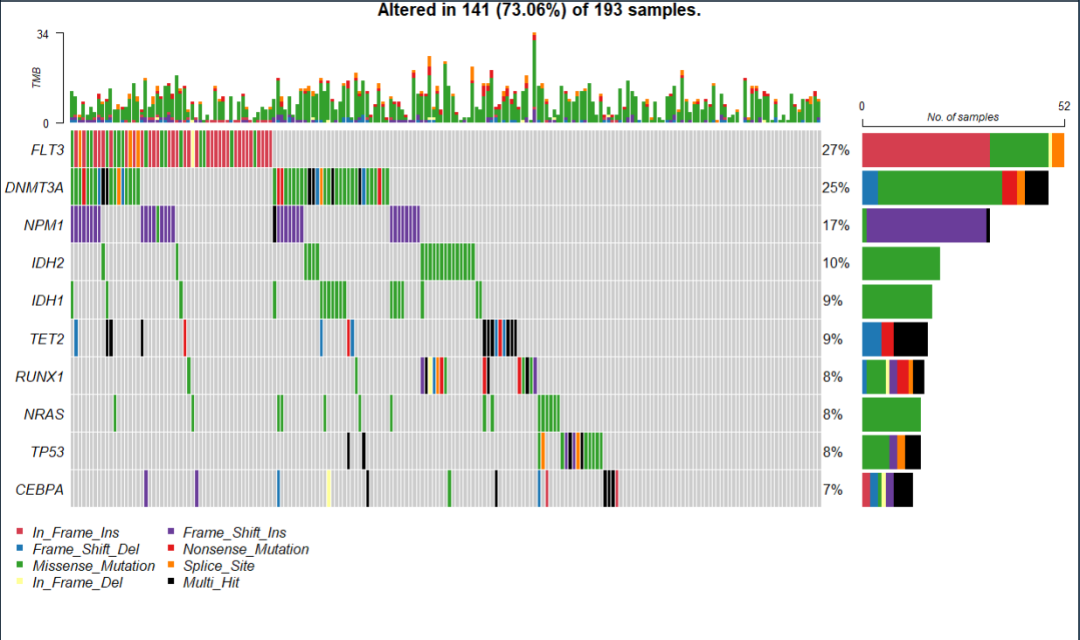
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |