一学就会!利用StringTie拼接转录本和定量(一)
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

小果之前带大家进行了转录组参考序列的比对,结果也是很顺利地跑出来了,接下来该做什么呢?当然可以进行基因表达水平分析啦。
进行基因表达水平分析可以帮助我们了解不同组织或条件下的基因表达情况,从而揭示生物体在不同生理或环境条件下的分子响应机制。
比如,在肿瘤研究中,我们可以通过分析肿瘤组织和正常组织的基因表达水平,找到肿瘤特异性基因并深入研究其调控机制,以期找到更好的肿瘤治疗方法;在植物研究中,我们可以通过分析在逆境条件下的基因表达情况,探索植物的应激响应机制并发现植物抗逆基因,以期提高作物的耐受性。因此,基因表达水平分析是分子生物学研究的重要手段之一。
重要性已经了解了,下面我们开始吧,在开始之前确保你已经拥有参考序列比对的结果哦:
我们需要用到samtools,如果没有的话,需要先安装:
###安装samtoolsconda create --name samtoolsconda activate circosconda install -c bioconda samtools
使用的时候记得激活一下环境。
我们先了解一下获得的sam数据,使用代码查看文件:
samtools view SRR8054374.sam | less
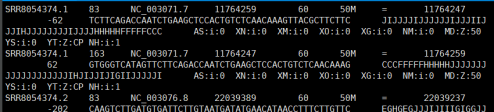
SRR8054374.1:序列 ID83:标记,标记了序列的比对情况,共有 16 种可能的取值,这里是 83NC_003071.7:参考序列 ID11764259:参考序列上比对位置的起点60:比对质量值,越高越可信50M:CIGAR 字符串,描述比对的匹配情况,M 表示匹配=:匹配到的参考序列的 ID,如果没有匹配则为 *11764247:参考序列上比对位置的终点-62:相对于序列起点的偏移量,注意是负数,表示反向互补链的偏移量TCTTCAGACCAATCTGAAGCTCCACTGTCTCAACAAAGTTACGCTTCTTC:序列数据JIJJJJIJJJJJIJJJIIJJJIHJJJJJJJJIJJJJHHHHHFFFFFCCC:序列的质量值,每个字符对应一个碱基的质量值AS:i:0:附加信息 AS,表示比对得分(Alignment Score),这里是 0XN:i:0:附加信息 XN,表示比对中的 N 的数量,这里是 0XM:i:0:附加信息 XM,表示比对中不同于参考序列的碱基数,这里是 0XO:i:0:附加信息 XO,表示比对中不同于参考序列的插入或删除的碱基数,这里是 0XG:i:0:附加信息 XG,表示比对中 G 的数量,这里是 0NM:i:0:附加信息 NM,表示比对中不同于参考序列的碱基数,这里是 0MD:Z:50:附加信息 MD,表示比对中不同于参考序列的碱基情况,这里是所有碱基都匹配YS:i:0:附加信息 YS,表示比对得分的第二种方式,这里是 0YT:Z:CP:附加信息 YT,表示比对类型,C 表示比对到参考序列的正向链,P 表示比对到参考序列的反向链NH:i:1:附加信息 NH,表示有多少个匹配到参考序列上的位置,这里是 1这个信息描述了一个序列在参考序列上的比对情况,可以根据这个信息来获取比对结果。接下来我们需要将结果转化为BAM文件,可以使用samtools软件包中的samtools view命令,这里小果使用循环语句批量转换格式:for file in *.samdonohup samtools view -bS $file > ${file%.sam}.bam &done
接下来等待结果即可!
格式转化成功后就可以进行下一步分析了!
小果今天就分享到这里,下期不见不散!
欢迎使用:云生信 – 学生物信息学 (biocloudservice.com)
如果想用服务器可以联系微信:18502195490(快来联系我们使用吧!)

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓