挖掘生物数据的隐藏模式?自组织映射SOM聚类一招解决!
{ 点击蓝字,关注我们 }
今天小师妹要给大家介绍的是一种常用于生物信息数据的聚类分析的方法——自组织映射(Self-Organizing Maps,SOM)。在基因表达谱数据分析中,SOM可以帮助研究人员发现基因表达的模式和关系,探索基因表达谱数据的生物学意义。在蛋白质结构分析中,SOM可以帮助研究人员理解蛋白质序列和结构之间的关系,并预测蛋白质的结构和功能。在基因组序列分析中,SOM可以帮助研究人员分析基因组序列之间的相似性和差异性,并识别基因组序列中的功能元件和重要区域。总之,SOM是一种强大的工具,可以帮助我们从海量的生物数据中挖掘出隐藏的模式和规律~那么现在就让小师妹带大家一起来学学吧~
SOM是一种无监督学习神经网络,可以将高维输入数据映射到低维空间中。其主要思想是将相似的输入数据映射到相邻的输出单元上,从而在低维空间中保留输入数据的拓扑结构信息。SOMs通常用于聚类、可视化和数据压缩等任务中。在SOM聚类中,每个单元格代表一个聚类中心,每个数据点都被分配到与其距离最近的聚类中心所在的单元格中。
SOM聚类的主要特点包括以下几个方面:
无监督学习:SOM聚类是一种无监督学习方法,不需要预先标记的训练数据。
高效性:SOM聚类算法具有高效性和可扩展性。由于它只需要在低维空间中计算数据点之间的距离,因此它可以处理大量的数据点。
保留拓扑结构:SOM聚类可以保留数据点之间的拓扑结构,这使得它能够捕捉数据中的更复杂的结构和模式。
可视化性:SOM聚类可以通过可视化工具将数据点投影到低维空间,以便更好地理解数据集的结构和模式。
SOM聚类的步骤通常包括以下几个方面:
数据预处理:首先需要对数据进行预处理,包括特征选择、特征缩放和数据清洗等。
创建SOM模型:使用SOM算法创建一个SOM模型,其中包括数据点的输入层、聚类中心的输出层和它们之间的连接权重。
训练SOM模型:使用训练数据集对SOM模型进行训练,以更新连接权重和聚类中心的位置,并不断调整模型以最小化损失函数。
可视化SOM模型:使用可视化工具(例如plot()函数)可视化SOM模型,以便更好地理解聚类的分布和特征。
识别聚类中心:对于每个聚类,通过查看其代表单元格的代码向量来理解其聚类中心的含义。
可视化聚类结果:使用可视化工具(例如ggplot2),将数据点可视化为SOM模型中的聚类,以帮助你理解聚类的分布和特征。
接下来小师妹将用一组组织基因表达数据带大家实践一下~
首先我们需要载入SOM相关的R包
library(kohonen)接下来就是载入我们的数据
library(tissuesGeneExpression)data(tissuesGeneExpression)
接下来我们就可以使用som( )函数进行模型的训练啦~
注意哦,som函数的data需要转换成矩阵的形式才能运行
e.matrix <- as.matrix(e[,1:95])该代码将训练一个5×5的SOM模型,其中rlen参数指定了训练迭代次数
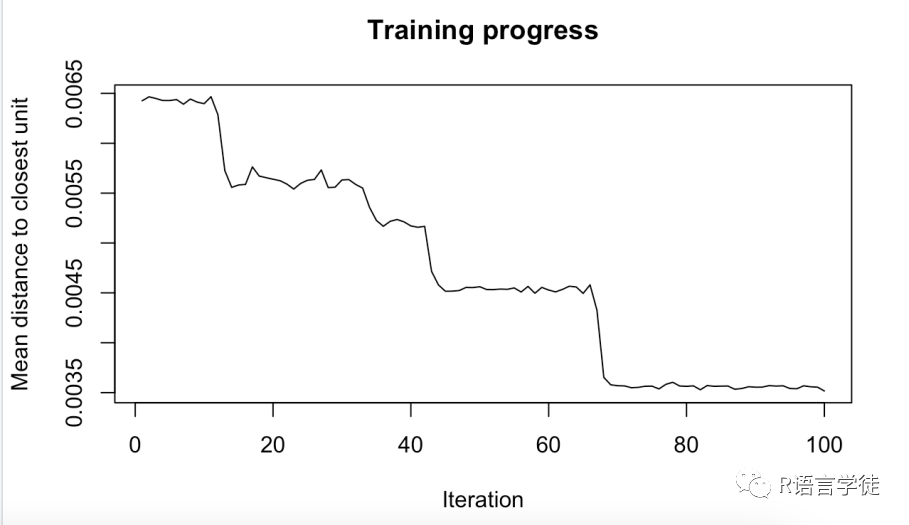
e.som <- som(e.matrix, grid = somgrid(5, 5, "hexagonal"), rlen = 100)plot(e.som, type = "changes")
使用type=“changes”可以展示训练过程,距离随着迭代减少的趋势,判断迭代是否足够;最后趋于平稳比较好
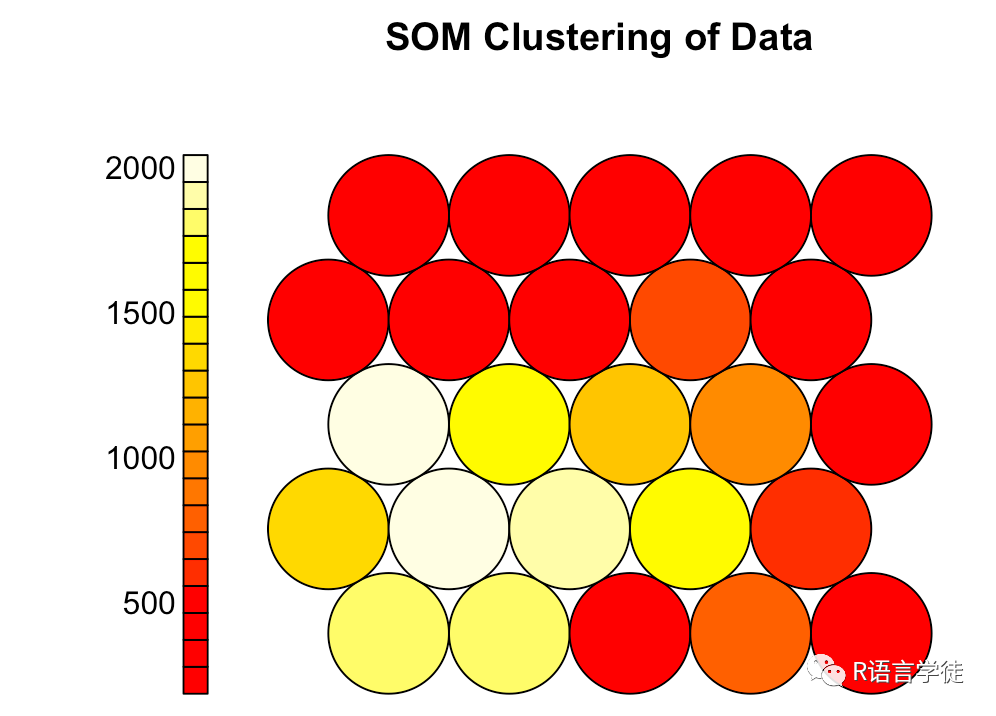
plot(e.som, type = “counts”, main = “SOM Clustering of Data”)使用type=“counts” 查看每个中心点包含的基因的数量
将type改成dist.neighbours来查看潜在的边界点,颜色越深表示与周边点差别越大,越是边界点
plot(e.som, type="dist.neighbours", main = "SOM neighbour distances", palette.name=grey.colors)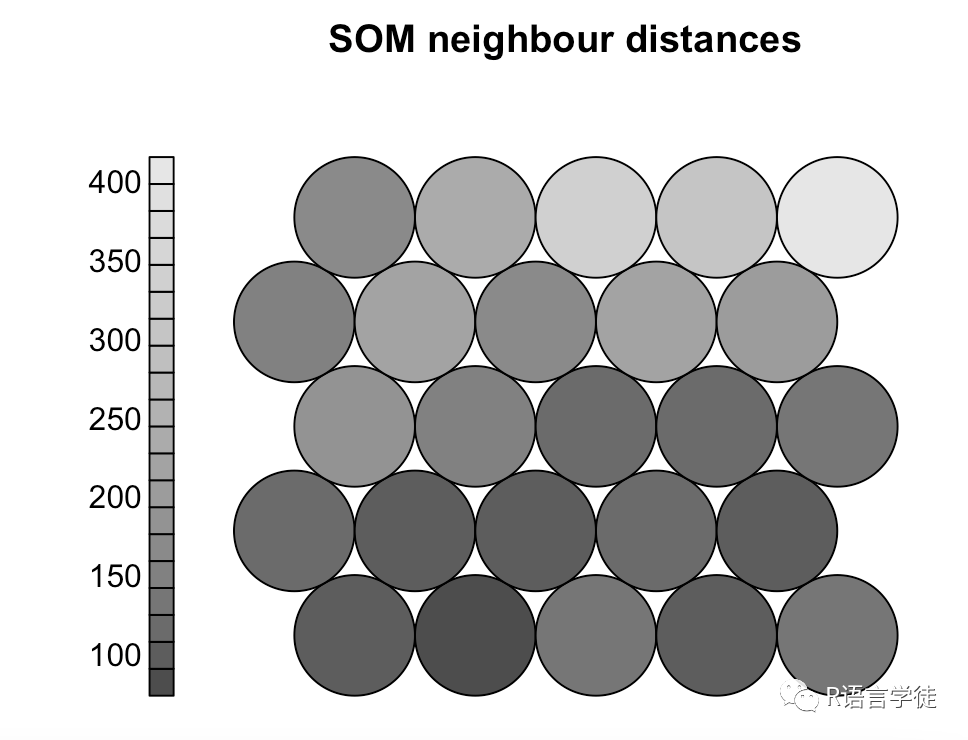
将type改成“codes”,查看中心点的变化趋势
plot(e.som, type = "codes", codeRendering="lines")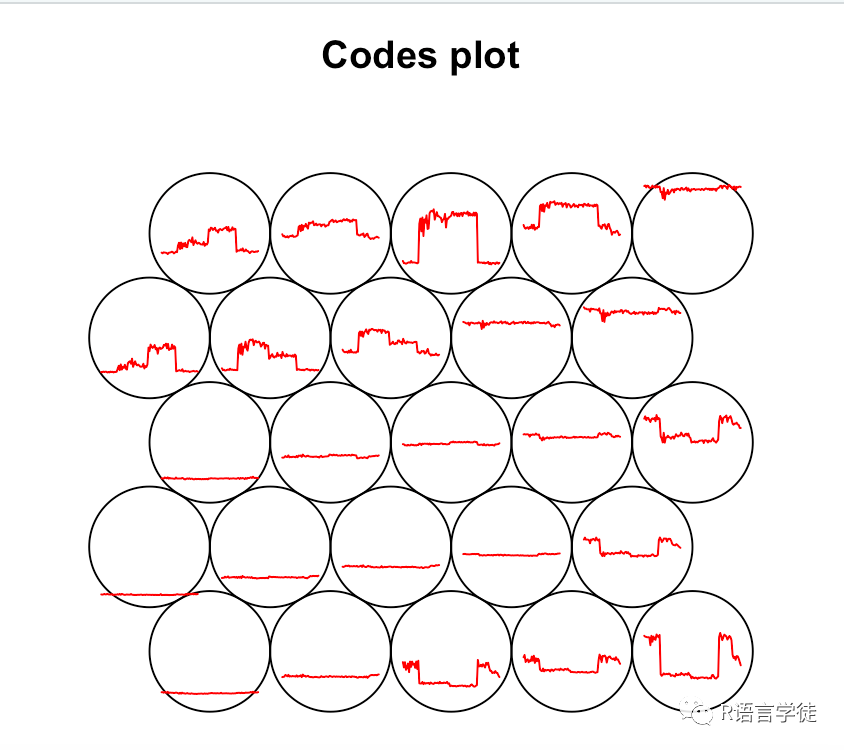
获取每个中心点相关的基因
table(e.som$unit.classif)
对结果进行进一步聚类
mydata <- as.matrix(as.data.frame(e.som$codes))wss <- (nrow(mydata)-1)*sum(apply(mydata,2,var))for (i in 2:15) wss[i] <- sum(kmeans(mydata, centers=i)$withinss)par(mar=c(5.1,4.1,4.1,2.1))plot(1:15, wss, type="b", xlab="Number of Clusters",ylab="Within groups sum of squares", main="Within cluster sum of squares (WCSS)")
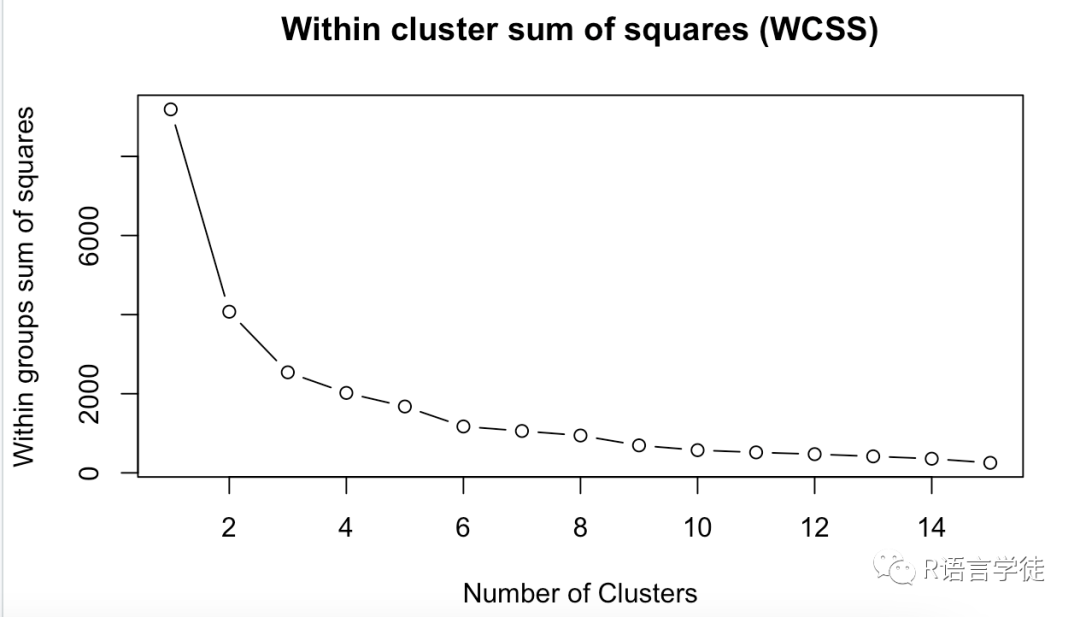
查看部分聚类结果
som_cluster <- cutree(hclust(dist(mydata)), 6)som_model_code_class = data.frame(name=rownames(e.matrix), code_class=e.som$unit.classif)som_model_code_class_cluster = som_model_code_classsom_model_code_class_cluster$cluster = som_cluster[som_model_code_class$code_class]head(som_model_code_class_cluster)
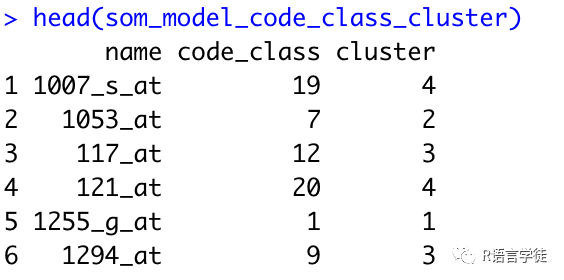
这就是SOM聚类的内容啦~看着这些神奇的可视化图像是不是很心动呢!赶紧和小师妹一起动手试试吧~
更多实用方便的小工具
在云生信平台等着你哦~
http://www.biocloudservice.com/home.html
E
N
D
