听说你还不会绘制回归分析模型中的森林图?
点击蓝字
关注小图
森林图
森林图(forest plot),从广义上来讲,它一般是在平面直角坐标系中,以一条垂直于X轴的无效线(通常坐标X=1或0)为中心,用若干条平行于X轴的线段,来表示每个研究的效应量大小及其95%可信区间,并用一个棱形来表示多个研究合并的效应量及可信区间,它是Meta分析中最常用的结果综合表达形式,现在也广泛应用在biomarker此类研究中。
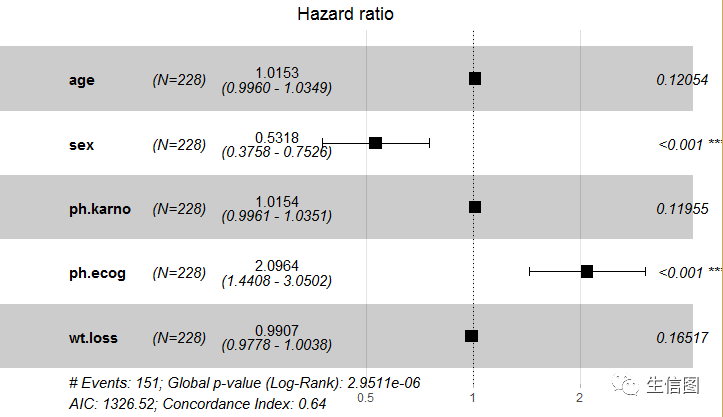
如下面图展示的森林图就是常规Cox回归结果图,主要显示了变量、病人数量、P值和HR值。比如: ph.ecog变量位于无效线(即中间的那条竖线)右侧,说明ph.ecog有助于死亡。森林图在常规情况下事件结局是”生/死”这种两分类,但有时候事件结局是”有效/无效”、”治疗/未治疗”等等其他二分类情况,评估事件是好事还是坏事。比如生存(生:0;死:1),位于无效线左侧的变量,说明这些变量不利于事件发生,是保护因素;位于无效线右侧的变量,说明这些变量有助于事件发生,是危险因素;当与无效线相交时,说明这些变量与事件发生之间关系不强!在整体数据上,用来评估这些变量因素对事件结局的影响。
绘制图片时首先构建构建COX回归模型
行为表达谱基因
列为样本的表达矩阵
expr <- read.table(file.path(data.path, " expr.txt"), header = T, sep = "t", row.names = 1,check.names = F,stringsAsFactors = F)expr <- expr[rowSums(expr > 0) > ncol(expr) * 0.1, ] 、
行为样本,列为结局信息的数据框
surv <- read.table(file.path(data.path, "surv.txt"), header = T, sep = "t", row.names = 1,check.names = F,stringsAsFactors = F)surv <- surv[surv$OS.time > 0, c("OS", "OS.time")] # 提取OScomsam <- intersect(rownames(surv), colnames(expr))expr <- expr[,comsam]; surv <- surv[comsam,,drop = F]
测试集
行为表达谱基因
列为样本的表达矩阵
T_expr <- read.table(file.path(data.path, "T_expr.txt"), header = T, sep = "t", row.names = 1,check.names = F,stringsAsFactors = F)生存数据是行为样本
列为结局信息
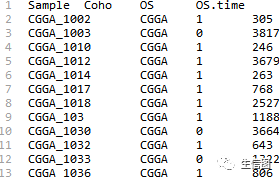
T_surv <- read.table(file.path(data.path, "T_surv.txt"), header = T, sep = "t", row.names = 1,check.names = F,stringsAsFactors = F)T_surv <- T_surv[T_surv$OS.time > 0, c("Coho","OS", "OS.time")] # 提取OScomsam <- intersect(rownames(T_surv), colnames(T_expr))T_expr <- T_expr[,comsam]; T_surv <- T_surv[comsam,,drop = F]
提取相同基因
comgene <- intersect(rownames(Train_expr),rownames(Test_expr))Train_expr <- t(Train_expr[comgene,]) # 输入模型的表达谱行为样本,列为基因Test_expr <- t(Test_expr[comgene,]) # 输入模型的表达谱行为样本,列为基因
模型
methods <- read.xlsx(file.path(code.path, "ESM.xlsx"), startRow = 2)methods <- methods$Modelmethods <- gsub("-| ", "", methods)head(methods)
训练集模型
model <- list()set.seed(seed = 123)for (method in methods){cat(match(method, methods), ":", method, "n")method_name = method #算法名称method <- strsplit(method, "\+")[[1]] #算法名称Variable = colnames(Train_expr) # 最后用于构建模型的变量for (i in 1:length(method)){if (i < length(method)){selected.var <- RunML(method = method[i], # 机器学习方法Train_expr = Train_expr, # 训练集有潜在预测价值的变量Train_surv = Train_surv, # 训练集生存数据mode = "Variable", #Variable(筛选变量)和Model(获取模型)timeVar = "OS.time", statusVar = "OS") # 用于训练的生存变量,必须出现在Train_surv中if (length(selected.var) > 5) Variable <- intersect(Variable, selected.var)} else {model[[method_name]] <- RunML(method = method[i],Train_expr = Train_expr[, Variable],Train_surv = Train_surv,mode = "Model",timeVar = "OS.time", statusVar = "OS")}}}
提到森林图,很多人的第一反应就是Meta分析。实际上,除了进行Meta分析以外,森林图还有很多的用处。森林图可以直观的反映出效应量(例如RR、OR、HR或者WMD)大小及其95% CI,这些效应量指标通常都是通过采用多因素回归分析所得,因此我们同样可以把森林图借鉴过来,用于展示单因素或者多因素回归分析的结果。
rt=read.table(file,header=T,sep="t",row.names=1,check.names=F)gene=rownames(rt)hr=sprintf("%.3f",rt$"HR")#获取95%置信区间#对HR (95% CI for HR)做处理,得到HR和low .95和high .95hrLow=sprintf("%.3f",rt$"HR.95L")hrHigh=sprintf("%.3f",rt$"HR.95H")Hazard.ratio=paste0(hr,"(",hrLow,"-",hrHigh,")")pVal=ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue))#保存图片pdf(file=outFile, width = 77 height =5)n=nrow(rt)nRow=n+1ylim=c(1,nRow)layout(matrix(c(1,2),nc=2),width=c(3,2))#森林图左边的基因信息xlim = c(0,3)par(mar=c(4,2,1.5,1.5))plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,xlab="",ylab="")text.cex=0.8text(0,n:1,gene,adj=0,cex=text.cex)text(1.5-0.5*0.2,n:1,pVal,adj=1,cex=text.cex);text(1.5-0.5*0.2,n+1,'pvalue',cex=text.cex,font=2,adj=1)text(3,n:1,Hazard.ratio,adj=1,cex=text.cex);text(3,n+1,'Hazard ratio',cex=text.cex,font=2,adj=1,)
绘制森林图
par(mar=c(4,1,1.5,1),mgp=c(2,0.5,0))xlim = c(0,max(as.numeric(hrLow),as.numeric(hrHigh)))plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,ylab="",xaxs="i",xlab="Hazard ratio")arrows(as.numeric(hrLow),n:1,as.numeric(hrHigh),n:1,angle=90,code=3,length=0.03,col="darkblue",lwd=2.5)abline(v=1,col="black",lty=2,lwd=2)boxcolor = ifelse(as.numeric(hr) > 1, 'red', 'blue')points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex=1.3)axis(1)dev.off()
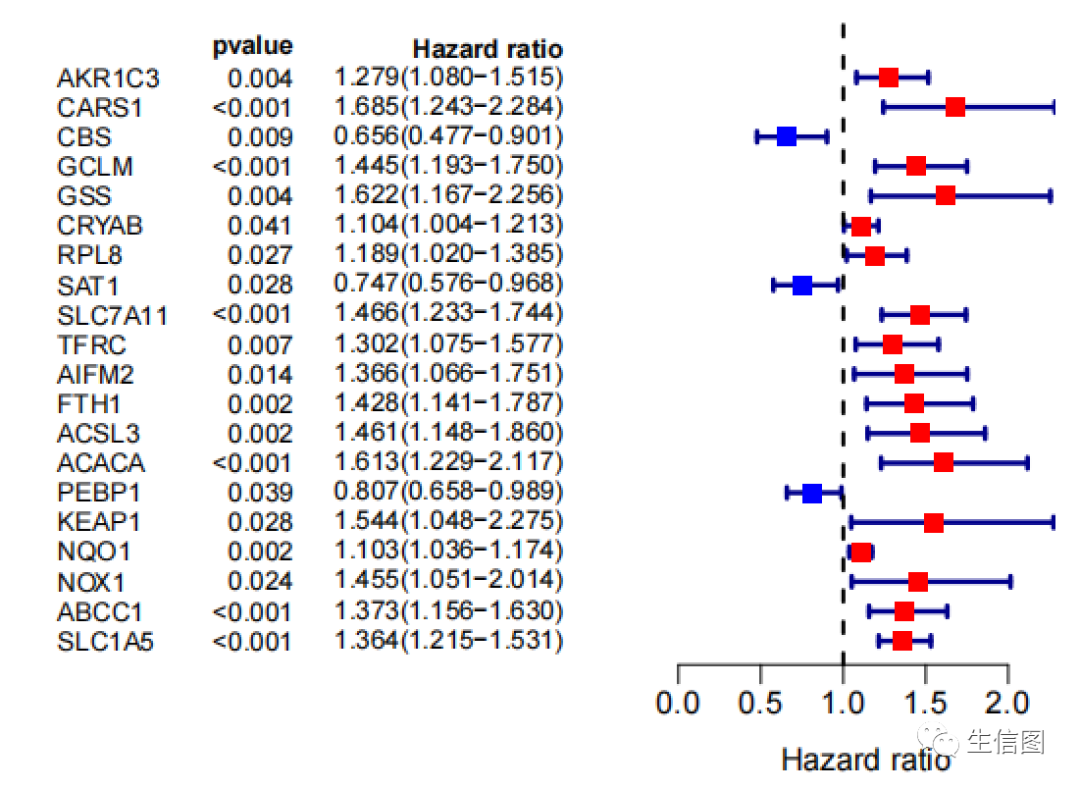
上面的绘图的思路和原理,应该也不难,大家可以自己练练手。
每一张科研制图都需要精雕细琢,勤写代码,多看帮助文档,可以让我们更好更快的掌握科研制图的精要,也能让我们的工作更完美的展现出来。加油吧!
今天的分享就到这里了,希望对大家有所帮助!

有疑问欢迎咨询小图的微信公众号(生信图)和云生信生物信息学平台( http://www.biocloudservice.com/home.html)
欢迎使用:云生信平台 ( http://www.biocloudservice.com/home.html)

|
往期推荐 |
|
|
|
|
|
|
👇点击阅读原文进入网址