师妹带你绘制不一样的临床相关分析热图
点
击
蓝
字
★
关
注
我
们
根据基因表达数据和临床数据生成热图,并可视化基因表达水平和临床性状之间的关联。其主要意义如下:
数据整合:代码将基因表达数据和临床数据进行整合,以便在后续分析中同时考虑基因表达水平和临床性状。
分组和排序:根据目标基因的表达量,将样本分为高表达组和低表达组,并根据基因表达水平对样本进行排序,以便后续可视化时能够呈现基因表达的模式。统计分析:代码使用卡方检验(chisq.test)来评估临床性状与基因表达水平之间的关联性,并根据检验结果给出显著性标记。这有助于研究人员识别哪些临床性状与基因表达存在显著关联。
热图绘制:代码使用ComplexHeatmap包绘制热图,将基因表达数据和临床性状的关联可视化。热图中,行表示样本,列表示基因和临床性状,颜色表示基因表达水平和临床性状的不同类别。结果展示:最终生成的热图可以帮助研究人员直观地了解基因表达水平和临床性状之间的关系。热图中的模式和颜色编码可以揭示基因表达的差异以及基因与临床性状之间的相关性。
#if (!requireNamespace("BiocManager", quietly = TRUE))# install.packages("BiocManager")#BiocManager::install("ComplexHeatmap")#BiocManager::install("limma")#引用包library(limma)library(ComplexHeatmap)expFile="geneExp.txt" #表达数据文件cliFile="clinical.txt" #临床数据文件setwd("C:\biowolf\Gene\14.cliHeatmap") #设置工作目录#通过library()函数引用了limma和ComplexHeatmap两个包。指定表达数据文件路径expFile和临床数据文件路径cliFile。#使用setwd()函数设置工作目录为"C:\biowolf\Gene\14.cliHeatmap"。通过read.table()函数读取表达数据文件和临床数据文件。提取表达数据文件中的基因名称和第一个基因的列数据。删除正常样本,只保留肿瘤样本的数据,并对重复样本求均值。根据目标基因的表达量将样本进行分组,并在数据框中添加一个名为"Type"的列。#读取表达数据文件rt=read.table(expFile, header=T, sep="t", check.names=F, row.names=1)gene=colnames(rt)[1]#删掉正常样品tumorData=rt[rt$Type=="Tumor",1,drop=F]tumorData=as.matrix(tumorData)rownames(tumorData)=gsub("(.*?)\-(.*?)\-(.*?)\-(.*?)\-.*", "\1\-\2\-\3", rownames(tumorData))data=avereps(tumorData)#根据目标基因表达量对样品进行分组Type=ifelse(data[,gene]>median(data[,gene]), "High", "Low")Type=factor(Type, levels=c("Low","High"))data=cbind(as.data.frame(data), Type)data=data[order(data[,gene]),]#读取临床数据文件,并对其中的"Age"列进行分类,根据年龄分为">65"和"<=65"两组。cli=read.table(cliFile, header=T, sep="t", check.names=F, row.names=1)cli[,"Age"]=ifelse(cli[,"Age"]=="unknow", "unknow", ifelse(cli[,"Age"]>65,">65","<=65"))

对临床性状进行循环,对每个临床性状与基因表达水平之间的关联进行卡方检验,得到显著性标记。将显著性标记添加到热图数据框rt的列名中。
#合并数据samSample=intersect(row.names(data), row.names(cli))data=data[samSample,"Type",drop=F]cli=cli[samSample,,drop=F]rt=cbind(data, cli)#对临床性状进行循环,观察临床性状在高低表达组之间是否具有差异,得到显著性标记sigVec=c(gene)for(clinical in colnames(rt[,2:ncol(rt)])){data=rt[c("Type", clinical)]colnames(data)=c("Type", "clinical")data=data[(data[,"clinical"]!="unknow"),]tableStat=table(data)stat=chisq.test(tableStat)pvalue=stat$p.valueSig=ifelse(pvalue<0.001,"***",ifelse(pvalue<0.01,"**",ifelse(pvalue<0.05,"*","")))sigVec=c(sigVec, paste0(clinical, Sig))}colnames(rt)=sigVec#定义了热图注释的颜色。基因表达水平使用蓝色和红色表示低和高,临床性状使用自定义的颜色向量表示。#rt=rt[apply(rt,1,function(x)any(is.na(match('unknow',x)))),,drop=F]bioCol=c("#0066FF","#FF9900","#FF0000","#ed1299", "#0dbc21", "#246b93", "#cc8e12", "#d561dd", "#c93f00","#ce2523", "#f7aa5d", "#9ed84e", "#39ba30", "#6ad157", "#373bbf", "#a1ce4c", "#ef3bb6", "#d66551","#1a918f", "#7149af", "#ff66fc", "#2927c4", "#57e559" ,"#8e3af4" ,"#f9a270" ,"#22547f", "#db5e92","#4aef7b", "#e86502", "#99db27", "#e07233", "#8249aa","#cebb10", "#03827f", "#931635", "#ff523f","#edd05e", "#6f25e8", "#0dbc21", "#167275", "#280f7a", "#6373ed", "#5b910f" ,"#7b34c1" ,"#0cf29a" ,"#d80fc1","#dd27ce", "#07a301", "#ddd53e", "#391c82", "#2baeb5","#925bea", "#09f9f5", "#63ff4f")colorList=list()colorList[[gene]]=c("Low"="blue", "High"="red")j=0for(cli in colnames(rt[,2:ncol(rt)])){cliLength=length(levels(factor(rt[,cli])))cliCol=bioCol[(j+1):(j+cliLength)]j=j+cliLengthnames(cliCol)=levels(factor(rt[,cli]))cliCol["unknow"]="grey75"colorList[[cli]]=cliCol}#绘制热图ha=HeatmapAnnotation(df=rt, col=colorList)zero_row_mat=matrix(nrow=0, ncol=nrow(rt))Hm=Heatmap(zero_row_mat, top_annotation=ha)#创建HeatmapAnnotation对象ha,用于定义热图的注释。空的矩zero_row_mat作为热图的主体数据。使用Heatmap()函数创建热图对象Hm,将注释对象ha作为顶部注释。#输出热图pdf(file="heatmap.pdf", width=7, height=5)draw(Hm, merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom")dev.off()#使用pdf()函数创建一个PDF文件,文件名为"heatmap.pdf",指定宽度和高度。使用draw()函数将热图对象Hm绘制到PDF文件中。
来给大家展示展示结果图吧,使用firefox命令。
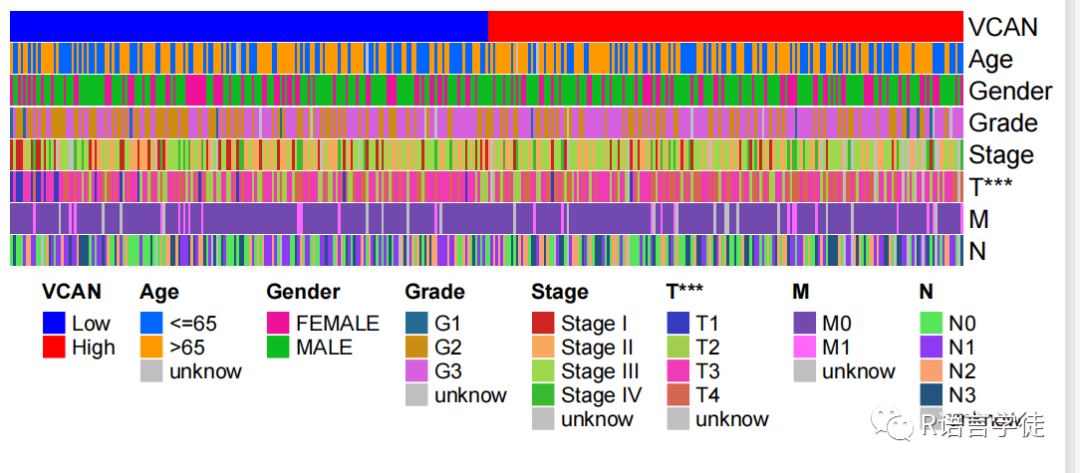
总之,综合分析基因表达数据和临床数据,并通过热图展示它们之间的关联。通过这种综合分析和可视化,可以更全面地理解基因表达和临床性状之间的关系,揭示潜在的生物学机制和指导进一步的研究。
下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:
http://www.biocloudservice.com/home.html。
★

