单细胞探秘:SingleCellExperiment揭开单细胞转录组学的神秘面纱!
点击蓝字 关注我们
hello,你知道如何使用SingleCellExperiment包来分析单细胞转录组学数据嘛?今天大海哥就来教大家如何具体使用这个R包,干货满满,感兴趣的话就和大海哥一起看下去吧!
走进SingleCellExperiment
首先,大海哥来简单的给大家介绍一下这个R包:
SingleCellExperiment包是用于存储和分析单细胞转录组学数据的R语言工具包。它提供了一个灵活的数据结构,可以容纳单细胞数据中的基因表达矩阵、元数据和其他相关信息,并为用户提供了多种功能来处理和分析这些数据。
SingleCellExperiment包的主要特点包括:
数据存储:SingleCellExperiment包使用一种称为SingleCellExperiment类的数据结构来存储单细胞数据。该类继承自SummarizedExperiment类,因此也可以使用SummarizedExperiment类的方法和函数。这个数据结构旨在简化单细胞数据的管理和访问。
灵活性:SingleCellExperiment包提供了灵活的数据存储方式。除了基因表达矩阵外,它还可以存储和管理降维数据、列数据、行数据和元数据。
数据分析:SingleCellExperiment包提供了许多用于处理和分析单细胞数据的函数和方法。它支持常见的数据操作,如子集选择、过滤、标准化和变换。
可扩展性:SingleCellExperiment包的数据结构可以方便地与其他单细胞数据分析工具和软件包进行交互。
总之,SingleCellExperiment包是一个功能强大且易于使用的工具包,专门设计用于处理和分析单细胞转录组学数据。它提供了灵活的数据存储方式和丰富的分析功能,使用户能够更好地理解和解释单细胞数据。
SingleCellExperiment进行单细胞数据分析
安装和加载 SingleCellExperiment 包
首先,我们需要确保已安装 SingleCellExperiment 包。如果尚未安装,请大家运行以下代码哦:
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("SingleCellExperiment")
然后,我们可以加载 SingleCellExperiment 包以开始使用:
library(SingleCellExperiment)创建 SingleCellExperiment 对象
1. 创建表达矩阵
首先,我们可以创建一个只包含表达矩阵的 SingleCellExperiment 对象。假设我们有一个 10×10 的表达矩阵,可以使用以下代码创建一个 SingleCellExperiment 对象:
counts <- matrix(rpois(100, lambda = 10), ncol=10, nrow=10)sce <- SingleCellExperiment(counts)
2. 加入降维数据
接下来,让我们创建一个包含降维数据的 SingleCellExperiment 对象。我们将使用 PCA 和 t-SNE 作为降维方法,并将它们存储在 reducedDims 中。以下是创建包含降维数据的 SingleCellExperiment 对象的示例:
ncells <- 100u <- matrix(rpois(20000, 5), ncol=ncells)v <- log2(u + 1)pca <- matrix(runif(ncells*5), ncells)tsne <- matrix(rnorm(ncells*2), ncells)sce <- SingleCellExperiment(assays=list(counts=u, logcounts=v),reducedDims=SimpleList(PCA=pca, tSNE=tsne))
3. 使用 SummarizedExperiment 对象转换
SingleCellExperiment 包还可以使用 SummarizedExperiment 对象转换生成 SingleCellExperiment 对象。以下是示例代码:
se <- SummarizedExperiment(assays=list(counts=u, logcounts=v))as(se, "SingleCellExperiment")
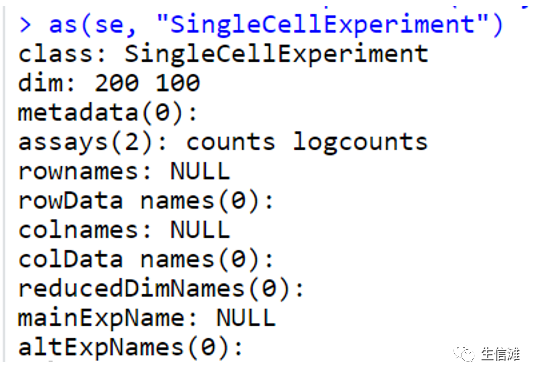
4. 含有 colData、rowData 和 metadata
此外,我们还可以添加列数据(colData)、行数据(rowData)和元数据(metadata)到 SingleCellExperiment 对象中。以下是一个示例,展示如何创建具有这些信息的 SingleCellExperiment 对象:
pretend.cell.labels <- sample(letters, ncol(counts), replace=TRUE)pretend.gene.lengths <- sample(10000, nrow(counts))sce <- SingleCellExperiment(list(counts=counts),colData=DataFrame(label=pretend.cell.labels),rowData=DataFrame(length=pretend.gene.lengths),metadata=list(study="GSE111111"))
提取行列信息
在 SingleCellExperiment 对象中,可以提取表达矩阵、元数据以及其他相关信息哦:
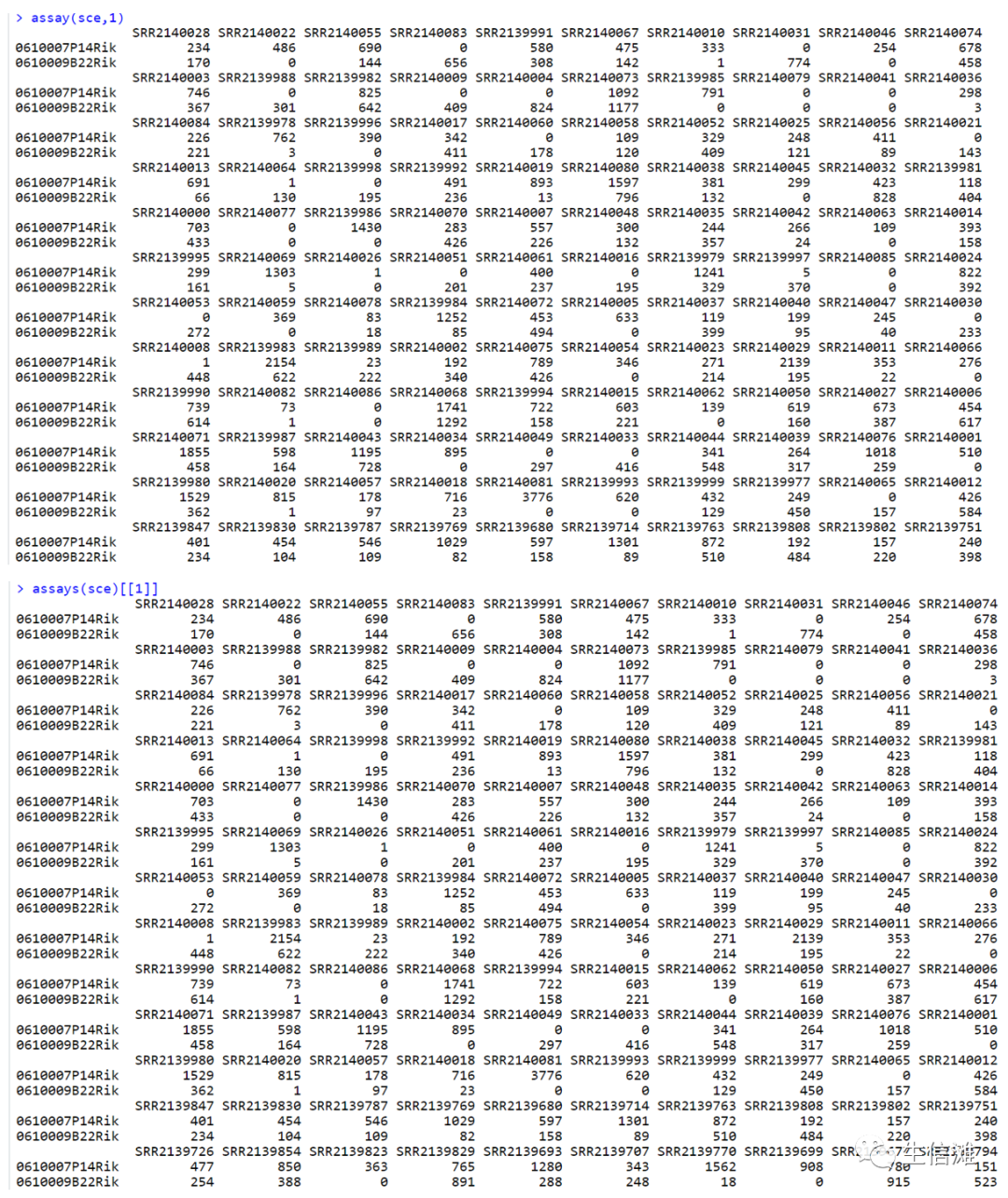
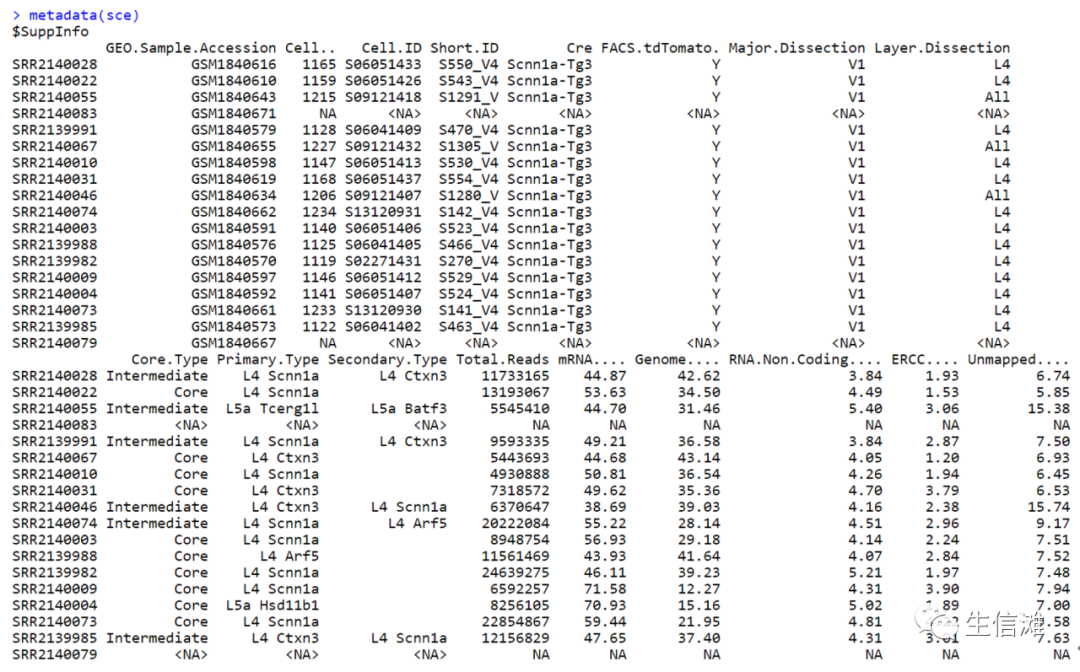
# 提取表达矩阵的维度dim(assay(sce))dim(sce)# 提取特定的表达矩阵assay(sce, 1)assays(sce)[[1]]# 提取列数据和行数据的名称colnames(colData(sce))rownames(rowData(sce))# 提取元数据metadata(sce)
特定的表达矩阵展示如下:
行数据和列数据如下:

元数据展示如下:
使用 Alternative Experiment 方法
SingleCellExperiment 包还提供了 Alternative Experiment 方法,用于处理不同形式的实验数据。以下是一些使用这些方法的示例:
# 查看可用的 Alternative Experiment 名称altExpNames(sce)# 访问指定的 Alternative ExperimentaltExp(sce, "ERCC")altExp(sce, 1)# 提取 Alternative Experiment 的表达矩阵assay(altExp(sce, "ERCC"))
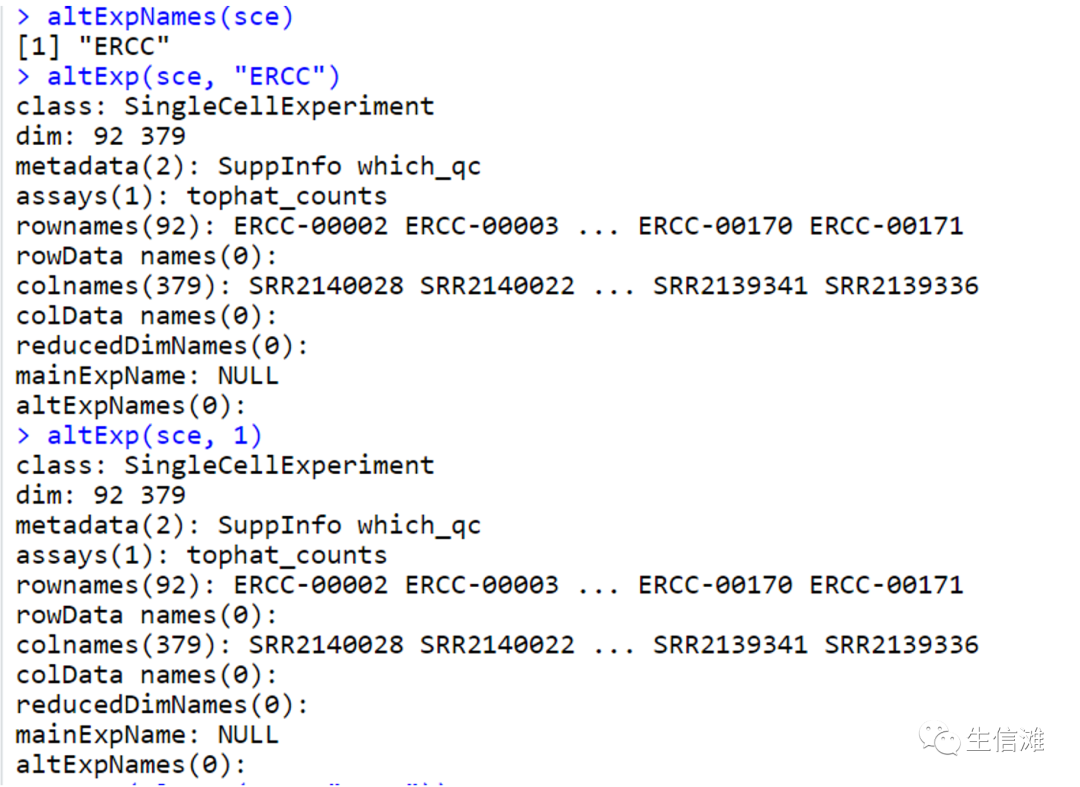
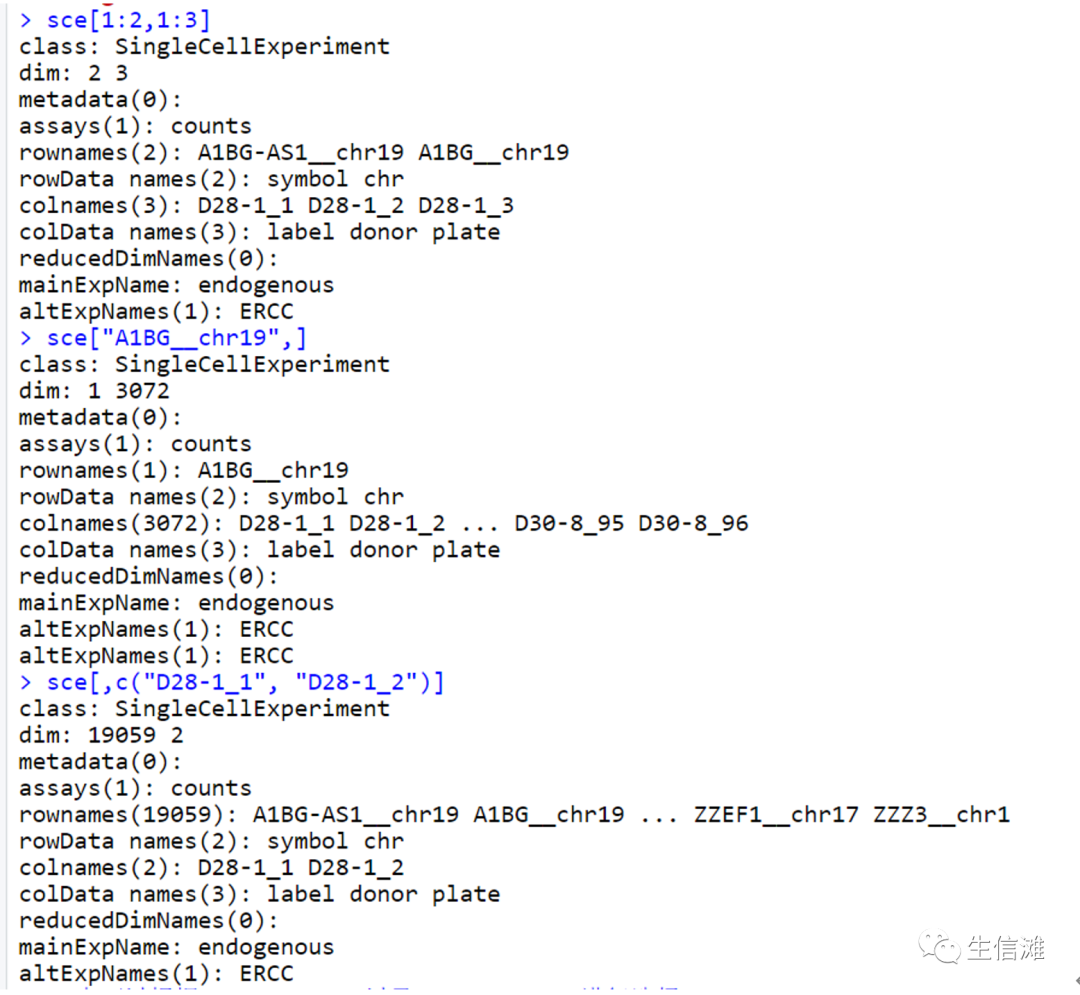
取子集
在 SingleCellExperiment 对象中,我们还可以根据指定的行和列来取子集。以下是示例代码:
# 使用索引取子集sce[1:2, 1:3]# 根据指定的行名取子集sce["A1BG__chr19", ]# 根据指定的列名取子集sce[, c("D28-1_1", "D28-1_2")]# 根据 rowData 和 colData 进行选择# 如果有 NA 值,需要去除sce[, sce$donore == "D28"]
运行结果展示如下:
以上就是SingleCellExperiment的基本使用方法,怎么样,今天的教程你学会了嘛?是不是很简单呢!更多学习干货要继续关注大海哥哦!


点击“阅读原文”进入网址