手把手教你可视化DESeq2 分析结果
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

上一节,小果向大家介绍了DESeq2差异分析的方法,DESeq2包也内置一些简单的可视化函数,只能满足我们的一些基础需求,如何将结果更丰富美观地进行可视化呈现呢?火山图(Volcano plot)和热图(Heatmap)是差异表达分析中常用的数据可视化工具,它们在结果解释和数据探索中起着重要的作用。下面接着跟小果一起来学习吧!
1.火山图
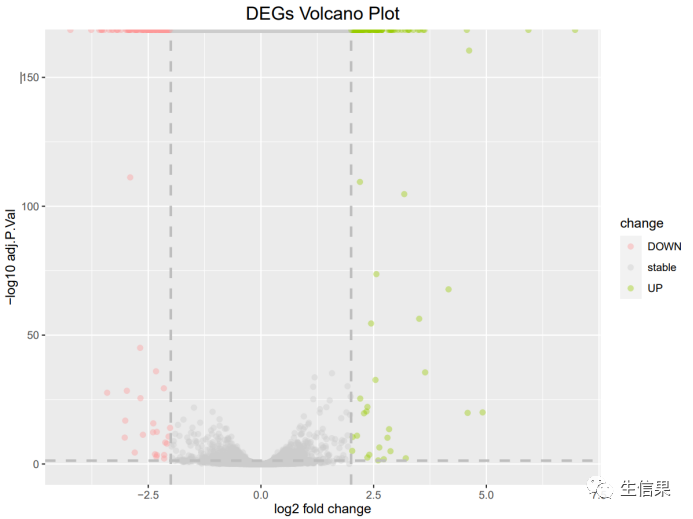
火山图是一种散点图,用于可视化差异表达基因的统计显著性和生物学重要性。在火山图中,横轴表示基因的差异表达水平(如折叠变化或对数变化),纵轴表示基因的统计显著性(如调整的 p-value)。每个基因以一个点的形式表示,点的位置取决于基因的差异表达程度和统计显著性。显著差异表达的基因通常位于图像的顶部或底部,而非显著差异表达的基因则位于图像的中央。通过观察火山图,研究人员可以快速识别出差异表达水平显著的基因,并了解它们的生物学重要性。
首先我们需要现设置筛选阈值,对于差异分析的结果来说,有两个比较重要的阈值,分别是矫正后的P值和log2FoldChange值。
矫正后的P值属于统计值阈值,在差异分析的过程中会采用公认的Benjamini-Hochberg校正方法对原有假设检验得到的显著性p值进行校正,一般选择0.01或者0.05作为标准。
log2FoldChange值是表达量的阈值,这个值代表两组样本间表达量的比值,一般将差异基因的筛选标准设置为log2FoldChange绝对值大于1或者2。
这两个指标的选取都是根据经验去进行设置,并不是完全固定,在具体分析过程中,我们通常需要根据得到的差异基因数目进行微调。
代码如下:
##设置筛选阈值logFoldChange = 2adjustP = 0.05diff_res <- as.data.frame(res)diff_res[is.na(diff_res)] <- 0
在火山图中我们可以看到,灰色的线就代表着这两个指标的阈值。这里我们将P值小于0.05并且log2FoldChange值大于2的基因定义为上调基因,将P值小于0.05并且log2FoldChange值小于-2的基因定义为下调基因。
diff_res$change <- ifelse(diff_res$padj > adjustP,'stable',ifelse(diff_res$log2FoldChange > logFoldChange,'UP',ifelse(diff_res$log2FoldChange < -logFoldChange,'DOWN','stable')))
接下来我们导入绘图所用R包ggplot2,设置图片名称。
#画火山图# 导入绘图包library(ggplot2)title <- paste("DEGs Volcano Plot", sep = "", collapse = "")
对应设置结果数据,x轴与y轴所绘制的数据,根据上一步所定义的上下调基因来绘制点的颜色。geom_hline与geom_vline函数用于绘制水平与垂直的辅助线。
ggplot(data=diff_res, aes(x=log2FoldChange, y=-log10(padj), color=change)) +geom_point(alpha=0.4, size=1.75) +theme_set(theme_set(theme_bw(base_size=20))) +xlab("log2 fold change") + ylab("-log10 adj.P.Val") +ggtitle(title) + theme(plot.title = element_text(size=15,hjust = 0.5)) +scale_colour_manual(values = c('#FF9999','gray80','#99CC00')) + #自定义颜色geom_hline(yintercept = c(-log10(adjustP)),linewidth = 1, color = "grey", lty = "dashed") +geom_vline(xintercept = c(-2,2),linewidth = 1, color = "grey", lty = "dashed") # 阈值辅助线
2.热图
热图是一种矩阵式的图形表示方法,用于可视化基因的表达模式。在热图中,每行代表一个基因,每列代表一个样本。通过对基因表达量进行归一化和聚类,热图展示了基因表达模式的相似性和差异性。热图中的颜色编码表示基因表达量的高低,通常使用不同颜色来表示高表达和低表达。热图可以帮助研究人员发现样本之间的相似性和差异性,识别出共同表达的基因模式,以及发现与特定条件相关的基因群。
首先我们根据上一小节得到的差异分析结果来获取差异表达的基因名称或ID。我们先根据p值对差异分析结果进行排序,使差异越大的基因排位越前。
diff_res <- diff_res[order(res$pvalue),]然后我们分别获取上调基因中排名前20的基因,与下调基因中排名前20的基因,并将其合并起来。这里可以根据需要设置所要展示的基因数量。
res_up_gene <- subset(diff_res,padj <= 0.05 & log2FoldChange >= 2)res_down_gene <- subset(diff_res,padj <= 0.05 & log2FoldChange <= -2)res_up_gene_top20 <- as.vector(rownames(head(res_up_gene,20)))res_down_gene_top20 <- as.vector(rownames(head(res_down_gene,20)))res_top40 <- c(res_up_gene_top20,res_down_gene_top20)
在我们获取到差异表达的基因后,我们可以通过热图更加直观地来呈现两组样本之间基因表达的差异。首先导入绘制热图所需要用到的R包“pheatmap”。
这里我们需要使用上一小节DESeq2包中我们构建的dds对象,获取dds对象中标准化后的counts 矩阵。
normalized_counts <- counts(dds,normalized=TRUE)这里我们选择前40个差异基因进行展示。
normalized_counts <- counts(dds,normalized=TRUE)然后直接调用pheatmap绘制热图,cluster_cols是设置是否进行列聚类,cluster_rows是是否进行行聚类,scale代表着数值是否进行标准化,可以规定按照行或者按照列来进行归一化。annotation_col是列的分组信息,对应地pheatmap包也具有设置行分组信息的参数annotation_row。
pheatmap(dat,cluster_cols =T,cluster_rows = T,scale="row", #校正,视图形而定annotation_col = coldata)dev.off()

今天小果给大家分享了两种可视化DESeq2分析结果的方式,火山图和热图在差异表达分析中起到了重要的作用,它们提供了一种直观和可视化的方式来理解差异表达基因的统计显著性和生物学相关性,可以帮助研究人员理解和解释差异表达基因的结果。小果的云生信平台上也提供绘制火山图(biocloudservice.com/406/406.php)和热图的工具(biocloudservice.com/747/747.php),使用起来非常方便快捷。更多学习资源请大家移步小果专属云生信平台(云生信 – 学生物信息学 (biocloudservice.com))搜索更多资源哦!
如果小伙伴有其他数据分析需求,可以尝试使用本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:(http://www.biocloudservice.com/home.html),其中也包括了通路表达分析(http://www.biocloudservice.com/313/313.php),单细胞的基因共表达分析(http://www.biocloudservice.com/906/906.php)等各种小工具哦~,有兴趣的小伙伴可以登录网站进行了解。
点击“阅读原文”立刻拥有
↓↓↓