想要探究微生物组学,却遇到了头疼的BIOM格式?别怕,带你用biomformat包打败BIOM格式,让你的科研之路畅通无阻!

公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240312
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~


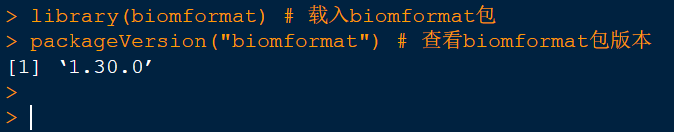
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")# 判断是否已经安装biomformat包BiocManager::install("biomformat")# 安装biomformat包,用于与BIOM格式交互输入library(biomformat) # 载入biomformat包packageVersion("biomformat") # 查看biomformat包版本显示如图所示版本号表示安装成功
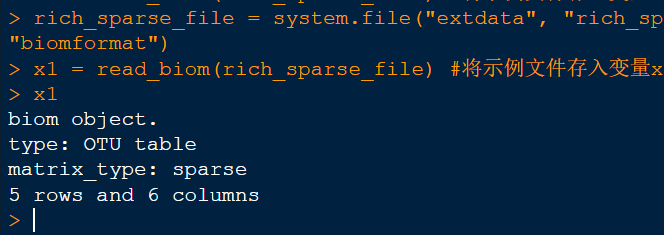
rich_sparse_file = system.file("extdata", "rich_sparse_otu_table.biom", package = "biomformat")x1 = read_biom(rich_sparse_file) #将示例文件存入变量x1x1 #显示x1文件类型
biom_data(x1)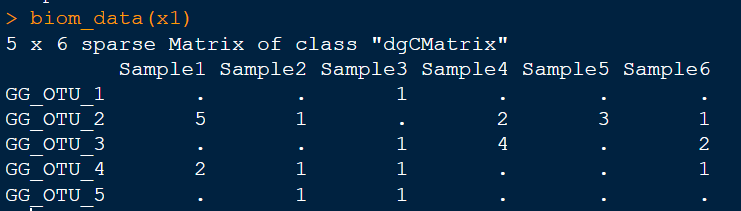
observation_metadata(x1)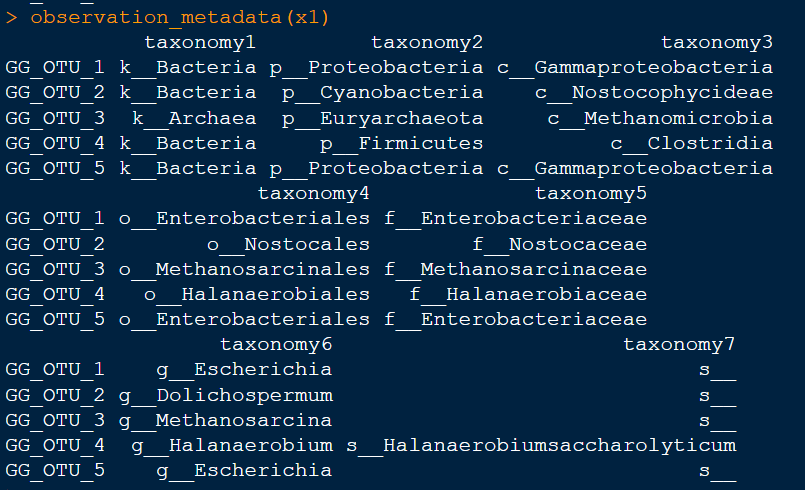
sample_metadata(x1)
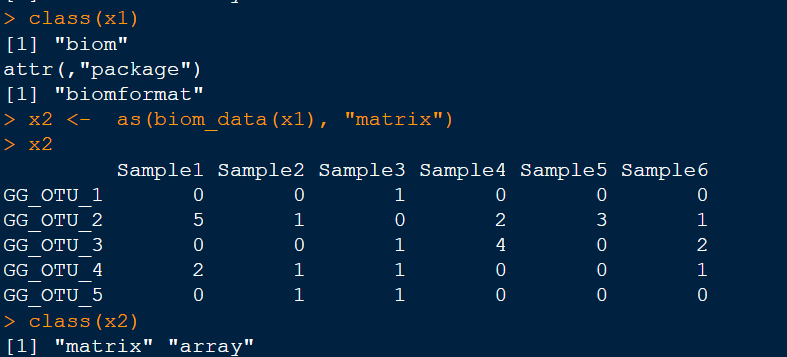
class(x1) #查看x1数据类型x2 <- as(biom_data(x1), "matrix") # 格式转换x2 # 查看x2内容class(x2) # 查看x2数据类型
plot(biom_data(x1)) # 点图 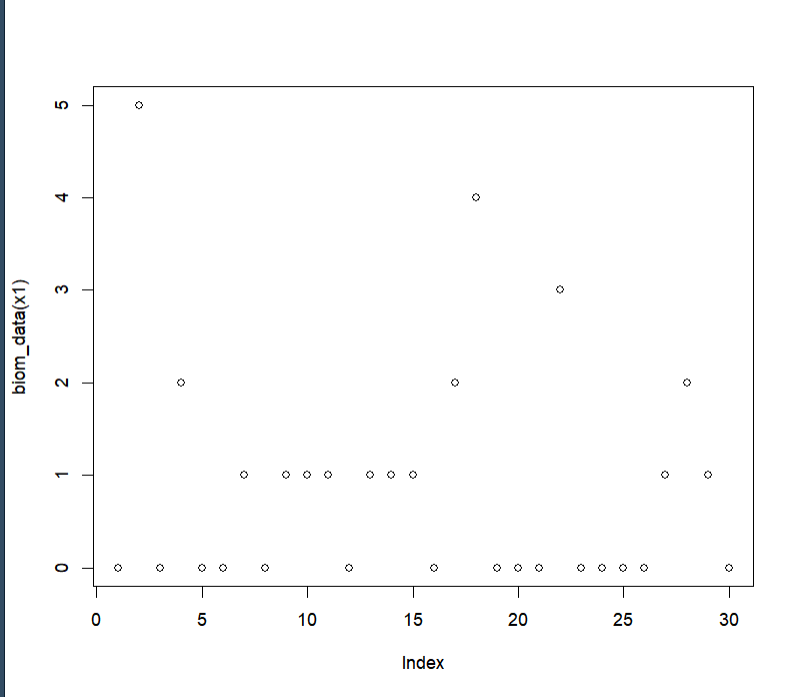
boxplot(as(biom_data(x1), "vector"), col = c("skyblue", "lightgreen"), pch = c(16, 18), xlab = "Group", ylab = "Values", main = "Boxplot of biomformat", width = 0.5, border = c("black", "black"), notch = TRUE, varwidth = TRUE) # 箱线图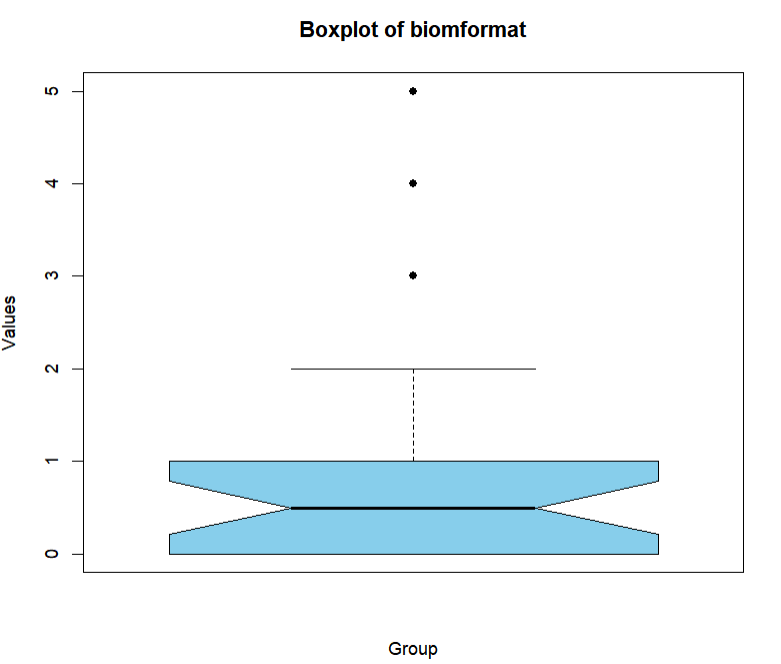
heatmap(as(biom_data(x4), "matrix")) 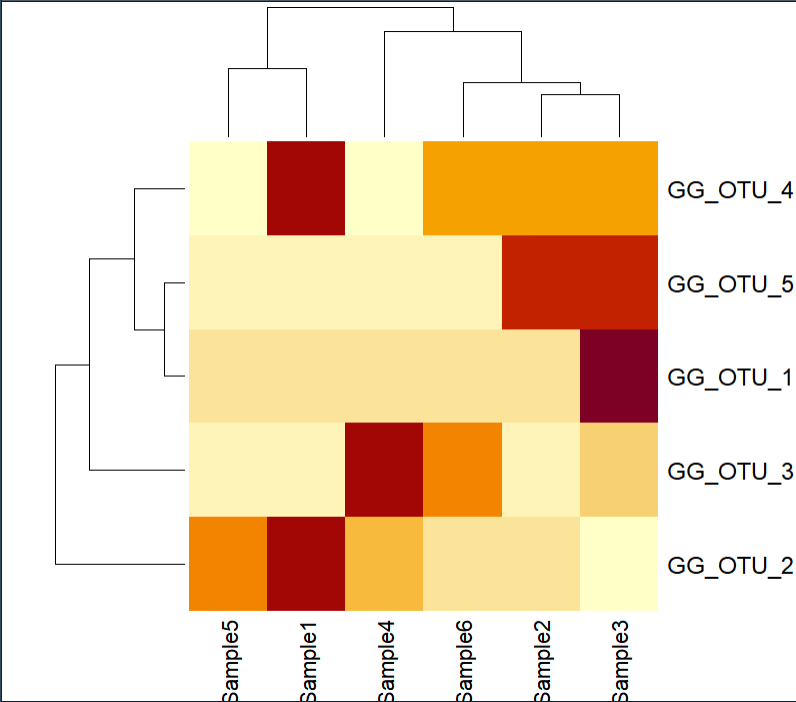
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |