李恒大神的又一杰作:bioawk让你轻松搞定生物序列分析


bioawk的安装

git clone git://github.com/lh3/bioawk.gitcd bioawmake或者connda安装::connda install -p ~/YOUR/conda path/ bioawk -y
bioawk的基本语法
bed:1:chrom 2:start 3:end 4:name 5:score 6:strand 7:thickstart 8:thickend 9:rgb 10:blockcount 11:blocksizes 12:blockstartssam:1:qname 2:flag 3:rname 4:pos 5:mapq 6:cigar 7:rnext 8:pnext 9:tlen 10:seq 11:qualvcf:1:chrom 2:pos 3:id 4:ref 5:alt 6:qual 7:filter 8:infogff:1:seqname 2:source 3:feature 4:start 5:end 6:score 7:filter 8:strand 9:group 10:attributefastx:1:name 2:seq 3:qual 4:comment
bioawk的常见应用
 如果我们想统计测序数据的基本信息,我们可以使用下面的命令来打印出每条序列的名称、GC比和长度:
如果我们想统计测序数据的基本信息,我们可以使用下面的命令来打印出每条序列的名称、GC比和长度: $ bioawk -c fastx '{ print $name, gc($seq),length($seq) }' test.fastq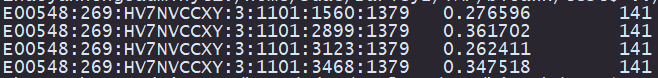
bioawk -c gff '{count[$3]++; len[$3] += $5 - $4 + 1} END {for (f in count) print f, count[f], len[f]}' test.gff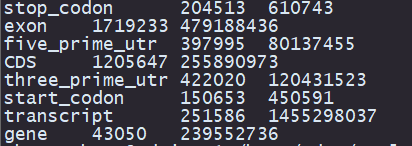
2. 过滤序列
$ bioawk -c fastx 'length($seq) >= 100 && meanqual($qual) >= 35' test.fastq > filtered_test.fastq
3. 转换格式
bioawk -c fastx -v OFS=',' '{print $name, length($seq), gc($seq)}' test.fastq > test.csv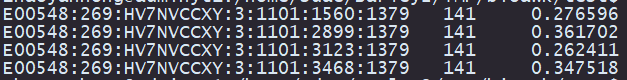
bioawk -c fastx '{ print ">"$name ORS revcomp($seq) }' test.fastq > rtest.fasta
samtools view test.bam | bioawk -c sam '{s=$seq; if(and($flag, 16)) {s=revcomp($seq)} print ">"$qname"n"s}' 总之,bioawk是一个非常强大的文本处理工具,它可以帮助我们快速地处理各种生物信息学相关的数据格式。它的语法简单易学,功能强大,可以让我们轻松地完成各种复杂的数据分析任务。如果您是一名生物信息学工作者,那么bioawk一定是您不可或缺的工具之一。希望这篇文章能够帮助你提高你的文本处理能力,让你在生物信息学中更加游刃有余。如果你想要了解更多关于bioawk的信息,你可以访问它的GitHub页面,或者查看它的手册。如果你有任何问题或者建议,欢迎在评论区留言或者联系我。谢谢你的阅读,下次再见。
总之,bioawk是一个非常强大的文本处理工具,它可以帮助我们快速地处理各种生物信息学相关的数据格式。它的语法简单易学,功能强大,可以让我们轻松地完成各种复杂的数据分析任务。如果您是一名生物信息学工作者,那么bioawk一定是您不可或缺的工具之一。希望这篇文章能够帮助你提高你的文本处理能力,让你在生物信息学中更加游刃有余。如果你想要了解更多关于bioawk的信息,你可以访问它的GitHub页面,或者查看它的手册。如果你有任何问题或者建议,欢迎在评论区留言或者联系我。谢谢你的阅读,下次再见。往期推荐

