TCGA数据库maf突变数据如何整合并绘制瀑布图,大海哥给你答案
点击蓝字 关注我们
小伙伴们好啊,我是大海哥,最近大海哥在知识的海洋里游荡时,发现了很多SCI文章中都有一种用于展示肿瘤细胞突变数据的图,大多都是下图这个模样。

于是大海哥又去查阅各类相关论文,发现大家都称其为体细胞突变(somatic mutation),其主要就是展示癌症体细胞的突变频率,同时可以根据这个数据计算肿瘤突变负荷(Tumor Mutation Burden, TMB),而要想画出这张图,首先我们必须要准本好数据。今天大海哥就先来跟大家聊聊怎么从TCGA数据库下载体细胞突变数据并计算TMB值。
首先我们先进入TCGA数据库主页(https://portal.gdc.cancer.gov/)。然后就可以在搜索框中输入我们感兴趣的癌症ID,如下图标记所示,大海哥就用常见的肺癌数据为例。

然后我们点击上面的检索推荐第一个,会出现下面的搜索结果页面,然后我们点击图示的红色标记处的Simple Nucleotide Variation的后面的571例case处

点进去后,我们发现有着很多种类的数据,然后我们需要一步一步筛选出我们需要的突变数据,需要大家按下图标记勾选,先点击Files,勾选上simple nucleotide variation、Masked Somatic Mutation、WXS、maf、open等选项,一个都不可以漏哦!

然后剩余的结果就是我们需要的数据,接着我们选择Case,点击Add all Files to Cart,
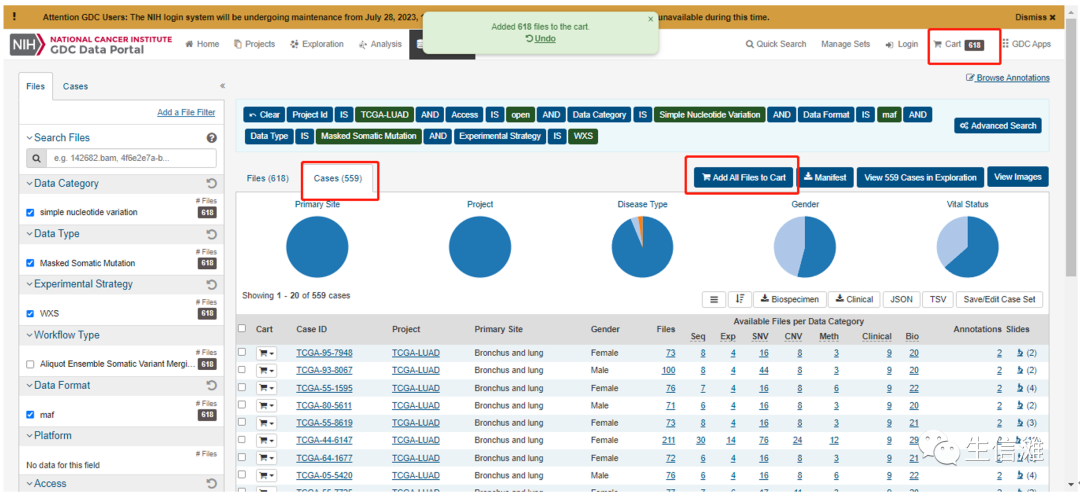
随后点击右上角的Cart,进去后,直接下载,选择Cart就可以啦!

然后我们会得到一个压缩包,我们需要把它解压放在文件夹中,会得到如图的文件
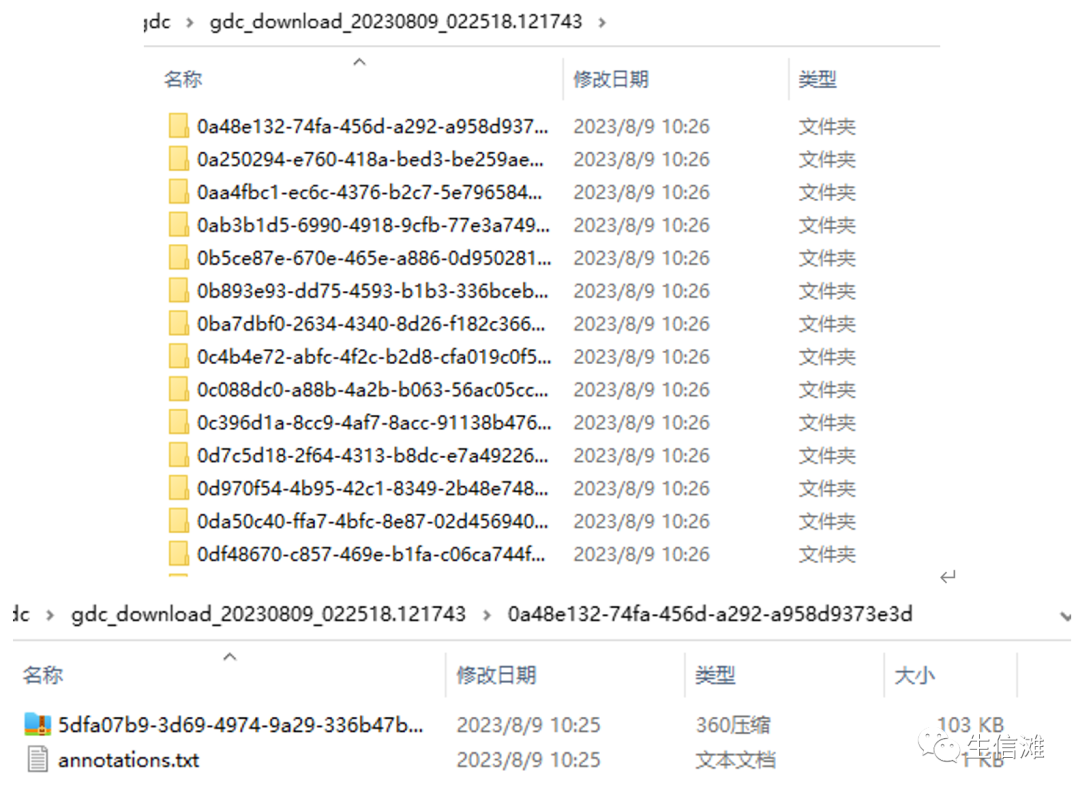
然后看着这么多文件夹,很多小伙伴肯定懵了,这怎么处理,手动整合?想想都害怕!别担心,大海哥来帮你,用代码一键解决!
#先设定工作目录,就设置为解压后的路径setwd("C:/Users/Administrator/Desktop/gdc/ gdc_download_20230809_022518.121743")#导入两个关键的R包library(maftools)library(dplyr)#获取所有的压缩文件,这一步可能会需要一些时间,取决于我们研究的癌症样本规模大小files <- list.files(pattern = '*.gz',recursive = TRUE)all_mut <- data.frame()for (file in files) {mut <- read.delim(file,skip = 7, header = T, fill = TRUE,sep = "t")all_mut <- rbind(all_mut,mut)}#数据整理all_mut <- read.maf(all_mut)a <- all_mut@data %>%.[,c("Hugo_Symbol","Variant_Classification","Tumor_Sample_Barcode")] %>%as.data.frame() %>%mutate(Tumor_Sample_Barcode = substring(.$Tumor_Sample_Barcode,1,12))gene <- as.character(unique(a$Hugo_Symbol))sample <- as.character(unique(a$Tumor_Sample_Barcode))mat <- as.data.frame(matrix("",length(gene),length(sample),dimnames = list(gene,sample)))mat_0_1 <- as.data.frame(matrix(0,length(gene),length(sample),dimnames = list(gene,sample)))for (i in 1:nrow(a)){mat[as.character(a[i,1]),as.character(a[i,3])] <- as.character(a[i,2])}for (i in 1:nrow(a)){mat_0_1[as.character(a[i,1]),as.character(a[i,3])] <- 1}#所有样本突变情况汇总/排序gene_count <- data.frame(gene=rownames(mat_0_1),count=as.numeric(apply(mat_0_1,1,sum))) %>%arrange(desc(count))# 大家可以修改数字20,代表TOP多少,也可选择自己感兴趣的gene_top <- gene_count$gene[1:20] ##保存save(mat,mat_0_1,file = "TMB.rda") ##保存为RDatawrite.csv(mat,"all_mut_type.csv")write.csv(mat_0_1,"all_mut_01.csv")#然后我们看一下保留的数据是什么样子的#all_mut_01.csv

# all_mut_type.csv
#终于获取到想要的数据了,然后就可以开始绘制瀑布图了oncoplot(maf = all_mut,top = 20, #显示前20个的突变基因信息fontSize = 0.6, #设置字体大小showTumorSampleBarcodes = F) #不显示病人信息
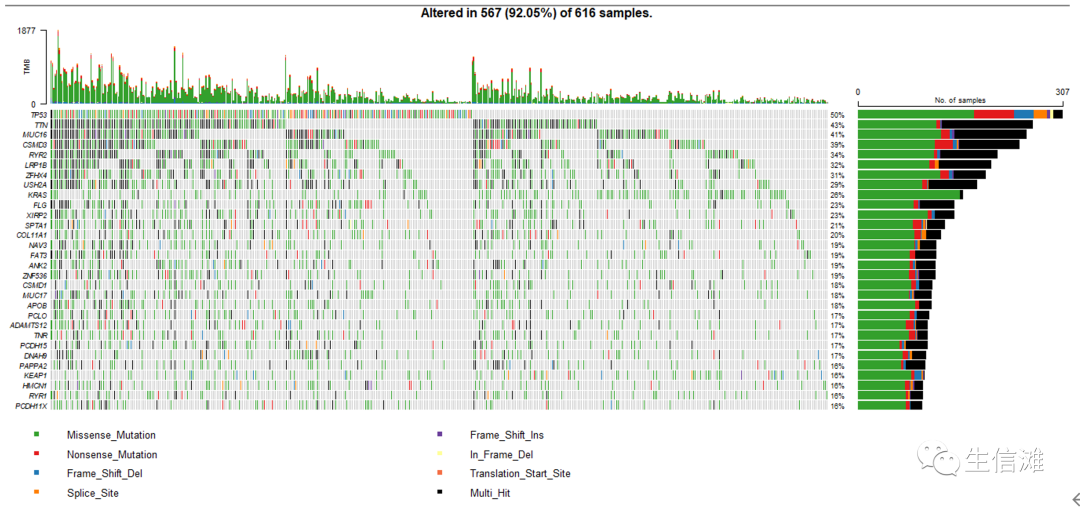
#大功告成#然后我们计算一下样本的TMB含量tmb_table = tmb(maf = all_mut) #默认以log10转化的TMB绘图tmb_table = tmb(maf = all_mut,logScale = F) #不logwrite.csv(tmb_table,"tmb_results.csv")#会同时绘制一个含量图

#看看导出的表格

#这个数据的total_perMB列就是我们需要的TMB含量


点击“阅读原文”进入网址