GWAS结果中SNP不会注释怎么办?Bedtools说好办!
点击蓝字
关注小图
Bedtools:一个强大的基因组算法工具集,由犹他大学昆兰实验室开发的。总的来说,Bedtools工具是一个堪称瑞士军刀的工具,用于广泛的基因组学分析任务。最广泛使用的工具使基因组运算成为可能:即基因组的集合论。例如,bedtools允许人们从广泛使用的基因组文件格式(如BAM、BED、GFF/GTF、VCF)的多个文件中交叉、合并、计数、补充和洗换基因组间隔。虽然每个单独的工具都被设计为执行相对简单的任务(例如,相交两个间隔文件),但是可以执行相当复杂的分析工作。
小图今天给大家介绍两种Bedtools安装方法
第一种
wget https://github.com/arq5x/bedtools2/archive/v2.25.0.tar.gztar xzvf v2.25.0cd bedtools2-2.25.0/makecd bin/
添加环境即可
export PATH=$PWD:$PATH第二种
使用conda安装
#创建自己的环境source activate mypy#进入自己的环境conda install -c bioconda bedtools
两种方法根据自己实际情况选择
首先,先用示例数据了解一下如何取数据集之间的交集基因
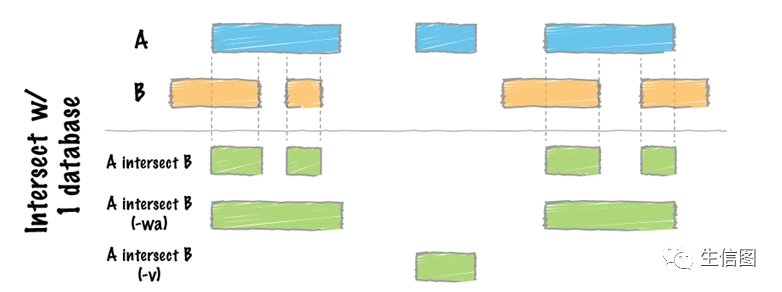
交集 -intersect
gene1.ped内容为

gene2.ped内容为

数据第1列:染色体
数据第2列:起始位置
数据第3列:终止位置
命令:
bedtools intersect -a gene1.ped -gene2.ped结果如下:
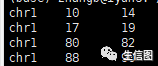
可见四个重复区域
第一个重复区域是10-14
第二个重复区域是17-19
第三个重复区域是80-82
第四个重复区域是88-90
如果想输出计算A中有B的交集区间,同时返回A中的结果:
可加 -wa 参数
命令:
bedtools intersect -a gene1.ped -gene2.ped -wa结果如下:
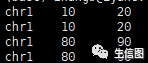
如果想输出计算A中有B的交集区间,同时返回B中的结果:
可加 -wb 参数
命令:
bedtools intersect -a gene1.ped -gene2.ped -wb结果如下:

从结果可见,加上-wb参数后,除了输出A中的交集区域外,还会输出B中的整个区间。
如果加上-wa -wb参数
命令:
bedtools intersect -a gene1.ped -gene2.ped -wa -wb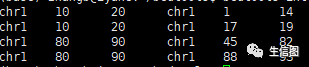
-c参数,统计A中每个区域与B重叠的次数
命令:bedtools intersect -a gene1.ped -gene2.ped -c

由结果可看出,在A文件中染色体位置和有多少B文件染色体位置与其有重叠。
那么,GWAS分析如何进行snp如何做注释,GWAS分析中,我们用基因型数据(SNP)+表型数据,进行关联分析,得到显著性的SNP,这些SNP有染色体和物理位置,那么我们如何对SNP进行基因注释呢?我们如何得到显著SNP附近的基因。
交集 -intersect
gene.ped内容为
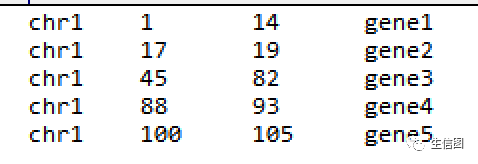
数据第1列:染色体
数据第2列:起始位置
数据第3列:终止位置
数据第4列:终止位置
snp.ped内容为

数据第1列:染色体
数据第2列:起始位置
数据第3列:终止位置
命令:
bedtools intersect -a snp.ped -b gene.ped -lojintersect,交集
-a,第一个位置信息表
-b,第二个位置信息表
-loj,以第一个为基准,返回结果

结果可以看到,第二个SNP区间,对应两个基因,写成了两行。第三个SNP区间没有对
应基因,用-1表示占位。共返回8行信息。
注意:每个SNP一行,如果有基因在其区间,放到右边,如果没有基因,返回空。如果一个SNP区间对应多个基因,写成多行。
如果不想要返回空值,只想返回有基因的SNP信息,命令如下:
bedtools intersect -a snp.ped -b gene.ped -wa -wb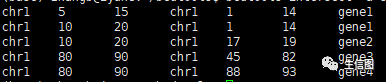
结果可以看到,没有匹配到基因的SNP都被删除了。

好了,今天的GWAS结果如何注释SNP就到这里了,希望小图的分享对大家的科研工作有所帮助。有兴趣的朋友可以继续关注小图的微信公众号(生信图)和零代码云生信生物信息学平台(http://www.biocloudservice.com/home.html)。小伙伴赶紧动手操作起来,看看今天的学习成果,说不定会成为你文章中的亮点,我们下期见~
欢迎使用:云生信平台 ( http://www.biocloudservice.com/home.html)

|
往期推荐 |
|
|
|
|
|
|
👇点击阅读原文进入网址