只需10分钟,搞定一套流程,GEO数据合并到去除批次效应
点击蓝字 关注我们
大家好啊,我是你们熟悉的大海哥,今天你大海哥要给大家分享的是一个流程教学,这个流程也是咱们生信人必须掌握的一个流程,但是很多小伙伴在这一步总是把握不好这个度,总是出现代码出问题不知道如何解决,那今天大海哥就做一个详细的R语言代码实现教学,让你彻底掌握如何整合GEO数据并去除批次效应的完整流程。
本次分析呢,我们选择大家最常见的CEL文件,那么CEL文件是什么呢?其实它的全称翻译就是探针的信号值和定位信息,是Affymetrix公司的芯片原始数据。
那么本次我们就是用同种疾病的两个数据集来完成这次分析教学,话不多说,开始吧!
首先,让我们打开GEO的数据集检索页面,
然后在右上角输入我们想要检索的数据集,例如大海哥找的这个数据集,

移到最下方,点击下载,然后就可以得到一个压缩包,我们首先需要把这个压缩包解压到一个文件夹中,然后就可以使用我们的代码来完成剩下的操作啦!
#首先加载需要的R包library(affy)library(limma)library(stringr)library(AnnoProbe)library(magrittr)#读取文件夹内的所有文件,data就是文件夹名,大家自行修改哦rawdata <- affy::ReadAffy(celfile.path = "data")#然后我们需要对数据进行rma标准化rawdata %<>% affy::rma()exprs <- Biobase::exprs(rawdata)range(exprs, na.rm = TRUE) # 1.889917 14.620563 不超过50不需要log2转化# 列重命名colnames(exprs) <- stringr::str_split(string=colnames(exprs),pattern = ".", simplify = T)[, 1]#然后一般我们都会选择箱型图查看一下数据的分布情况boxplot(exprs, outline = FALSE, notch = FALSE, las = 2)

#同时我们还可以对其进行limma标准化exprs %<>% limma::normalizeBetweenArrays()boxplot(exprs, outline = FALSE, notch = FALSE, las = 2)range(exprs, na.rm = TRUE) # 2.09520 14.30741exprs %<>% as_tibble(rownames = "probe_id")

#然后我们需要获取对应的探针注释文件# 得到探针对应的基因名字probe2Symbol <- AnnoProbe::idmap("GPL570")# 看看前几行head(probe2Symbol,3)

然后转换一下探针的ID为Symboltransid <- function(probe2Symbol, exprs, method = "median") {probe2Symbol$probe_id %<>% as.character()exprs$probe_id %<>% as.character()exprs %>%dplyr::inner_join(probe2Symbol, by = "probe_id") %>%dplyr::select(-probe_id) %>%dplyr::select(symbol, everything()) %>%dplyr::mutate(ref = apply(across(where(is.numeric)), 1, method)) %>%dplyr::arrange(desc(ref)) %>%dplyr::select(-ref) %>%dplyr::distinct(symbol, .keep_all = TRUE)}expression <- transid(probe2Symbol, exprs, method = "median")
#然后我们这就得到了一个整理好的数据#接下来我们需要找到另一个数据集,然后开始合并,这里就不介绍相同的流程啦!直接快进到合并环节吧!#没有请先安装这个包BiocManager::install("sva")#导入数据,count数据就是我们合并后的数据,保留相同基因然后直接合并哦,count <- read.csv("count.csv",header = T,row.names = 1)#过滤掉低表达的基因Expr <- count[rowSums(count)>1,]##载入样品信息,含批次data <- read.table("data.txt",header = T)data[,2] <- as.factor(data$type)##type表示的是生物学上的不同处理data[,3] <- as.factor(data$batch)##batch表示不同的批次信息row.names(data) <- data$sample

#batch对应的就是样本的批次,batch1就是第一个数据集,batch2就是第二个数据集#我们先来看看处理之前,数据的分布情况library(FactoMineR)##没有请先安装pre.pca <- PCA(t(Expr),graph = FALSE)fviz_pca_ind(pre.pca,geom= "point",col.ind = data$batch,addEllipses = TRUE,legend.title="Group" )
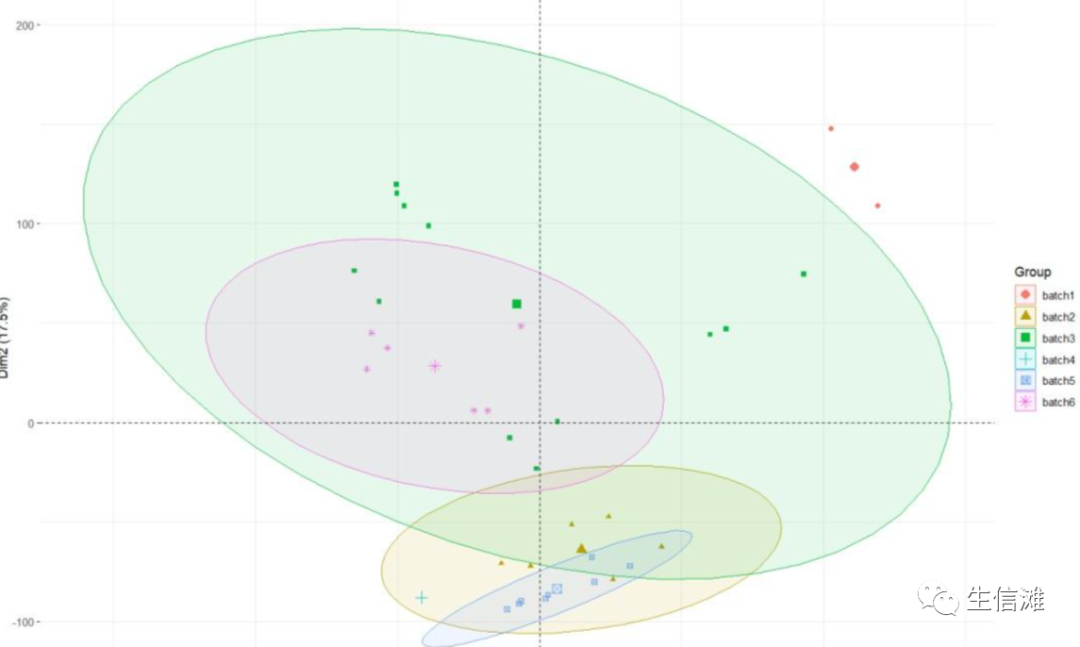
#可以看到处理之前,数据都是在不同的区域,说明存在一定的批次效应#建立批次效应的模型,data$type表示的是数据中除了有不同的批次,还有生物学上的差异。#在校正的时候要保留生物学上的差异,不能矫正过枉。model <- model.matrix(~as.factor(data$type))##消除批次效应library(sva)combat_Expr <- ComBat(dat = Expr,batch = data$batch,mod = model)##combat_Expr就是校正后的数据#再看看处理之后的数据library(FactoMineR)af1.pca <- PCA(t(combat_Expr),graph = FALSE)combat.pca <- PCA(t(combat_Expr),graph = FALSE)fviz_pca_ind(combat.pca,geom= "point",col.ind = data$batch,addEllipses = TRUE,legend.title="Group" )

#可以看到,不同批次的样品重叠在一起,证明批次效应可以通过sva包降低。#接着就可以用combat_Expr 这个数据做后续分析啦!#虽然但是,可以打开这份校正后的数据看看,你会发现似乎有一些数据出现了负数的情况,而且本来为整数的count数据也变成了小数。这样就不能用DESeq2包进行差异分析了,因为这个包要求的基因表达量数据必须为整数的row_count数据,所以后续差异分析可以使用limma包来完成哦!


点击“阅读原文”进入网址