snakemake–最好用的流程管理工具之一,你值得拥有
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

-
还在对着实验室上古大佬留下脚本发愁吗?还在头疼大shell套小shell的屎山吗?还在为找不到问题步骤的shell而烦恼吗?快跟着小果把snakemake学起来吧。

Snakemake的安装
首先当然是创建一个snakemake的文件夹啦,文件管理还是很重要滴。
mkdir snakemake
cd snakemake
然后下载解压官方的示例文档
curl -L https://api.github.com/repos/snakemake/snakemake-tutorial-data/tarball -o snakemake-tutorial-data.tar.gztar --wildcards -xf snakemake-tutorial-data.tar.gz
看到那个environment.yaml了吗,那就是教员佛祖真主赐予我等菜鸡救赎,

Snakemake连运行环境都帮我们配置好了,github下载的大神pipeline直接运行mamba(不要慌,mamba是conda的强力替代品)
mamba env create --name snakemake --file environment.yamlconda activate snakemake
官方示例文件结构如下图所示

接下来就到了激动人心的流程搭建了
rule bwa_map: #定义第一条规则,命名为bwa_mapinput:"data/genome.fa","data/samples/A.fastq"output:"mapped_reads/A.bam"shell:"bwa mem {input} | samtools view -Sb - > {output}"
当然,实际项目怎么可能只有一个样本,我们就不能直接写样本名了,snakemake支持使用通配符来批量运行命令,修改一下上面的代码,当然,sample可以做个yaml文件,或者在shell里写个for,这里容小果偷个懒
rule bwa_map: #定义第一条规则,命名为bwa_mapinput:"data/genome.fa","data/samples/{sample}.fastq"output:"mapped_reads/{sample}.bam"shell:"bwa mem {input} | samtools view -Sb - > {output}"

流程流程,当然不能只有一个规则啦。接下来再写个sort规则,在头文件把sample定义了,小样本测试可以这样偷懒,大样本还是要yaml文件哦
sample = [“A”, “B”, “C”]rule bwa_map: #定义第一条规则,命名为bwa_mapinput:"data/genome.fa","data/samples/{sample}.fastq"output:"mapped_reads/{sample}.bam"shell:"bwa mem {input} | samtools view -Sb - > {output}"rule samtools_sort:input:"mapped_reads/{sample}.bam"output:"sorted_reads/{sample}.bam"shell:"samtools sort -T sorted_reads/{wildcards.sample} ""-O bam {input} > {output}"rule samtools_index:input:"sorted_reads/{sample}.bam"output:"sorted_reads/{sample}.bam.bai"shell:"samtools index {input}"
现在,见证奇迹的时刻
snakemake --dag sorted_reads/{A,B}.bam.bai | dot -Tsvg > dag.svg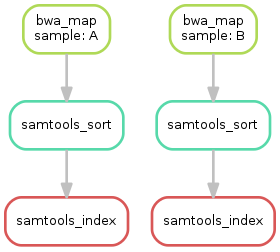
这才是snakemake的精髓有木有,直接生成可视化的流程图,妈妈再也不怕小果看流程shell一脸懵了,天下就没有我小果看不懂的流程。
好啦,不多说了,今天的内容就到这里了,你学会了吗!

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓