注意了!注意了!三分钟手把手教你用ape包画出漂亮美观的系统发育树!

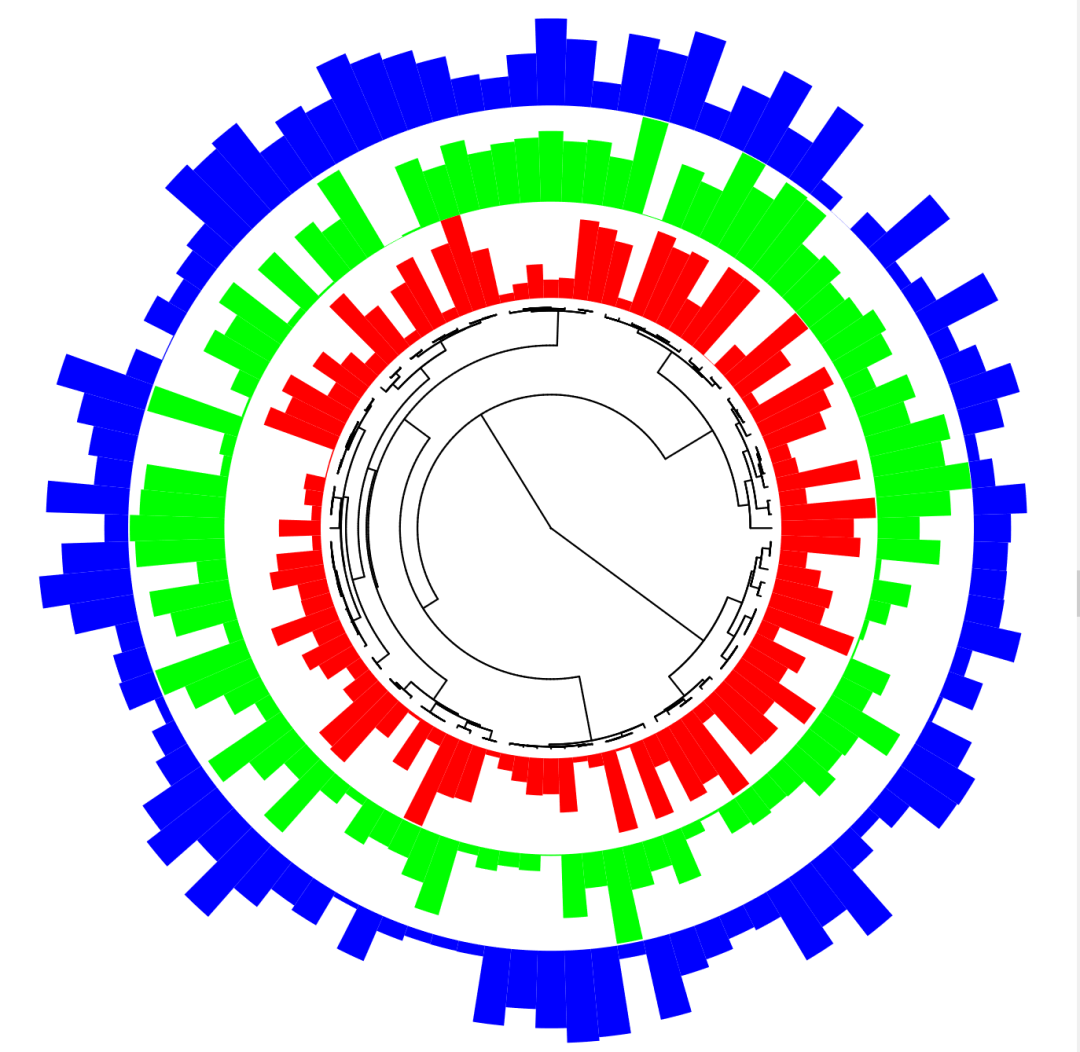
同学们一定都知道,系统发育树是生物学研究中非常重要的工具,在生物学研究中具有广泛的应用价值,它可以帮助我们研究物种的进化历程、推测共同祖先和演化路径。那我们怎样自己动手画出像上图一样美观的系统发育树呢?今天小果将向大家介绍一个功能强大的R包——ape包,它不仅为我们提供了非常多用于构建、比较和可视化系统发育树的函数和方法,同时还支持绘制不同类型的系统发育树,如进化树、无根树和圆形树等,以满足不同研究需求。小果将带领大家学习从ape包的安装到绘制不同类型系统发育树,再到自定义叶节点属性的全部过程,通过本篇对ape的学习,你就能像小果一样,动动手,三分钟画出漂亮美观的系统发育树。通过对ape包的学习,我们可以更好地理解和解释系统发育树的结果,进一步探索和应用系统发育树在生物学研究中的重要性和价值。接下来,就让小果带领大家正式开启对ape包的学习之旅吧!
公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240423
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

if (!require("BiocManager", quietly = TRUE))install.packages("ape")BiocManager::install("ape") # 在BiocManager环境下安装ape查看是否安装成功packageVersion("ape") # 查看DNAcopy版本

library(ape) # 载入ape包mytr <- read.tree(text = "((Pan:5,Homo:5):2,Gorilla:7);") # 把示例文件转化为树类紧接着,我们需要建立一个函数,用来在树的周围显示坐标边框,并且把x和y轴都设置为绿色:foo <- function() {col <- "green"for (i in 1:2)axis(i, col = col, col.ticks = col, col.axis = col, las = 1)box(lty = "19")} # 创建函数用来绘制坐标框接下来,我们以四种不同的方式对树进行绘图,并都调用foo函数,我们可以使用layout()函数来划定绘图区域,在一个大图上同时绘制四个系统发育树来更直观的展示ape包的功能。layout(matrix(1:4, 2, 2, byrow = TRUE)) # 划分绘图区域plot(mytr); foo() # 默认参数绘制系统发育树plot(mytr, "c", FALSE); foo() #绘制进化系统发育树plot(mytr, "u"); foo() # 绘制无根系统发育树par(xpd = TRUE) # 允许图形超过边框plot(mytr, "f"); foo() # 绘制圆形系统发育树box("outer") # 外部再绘制一个大边框
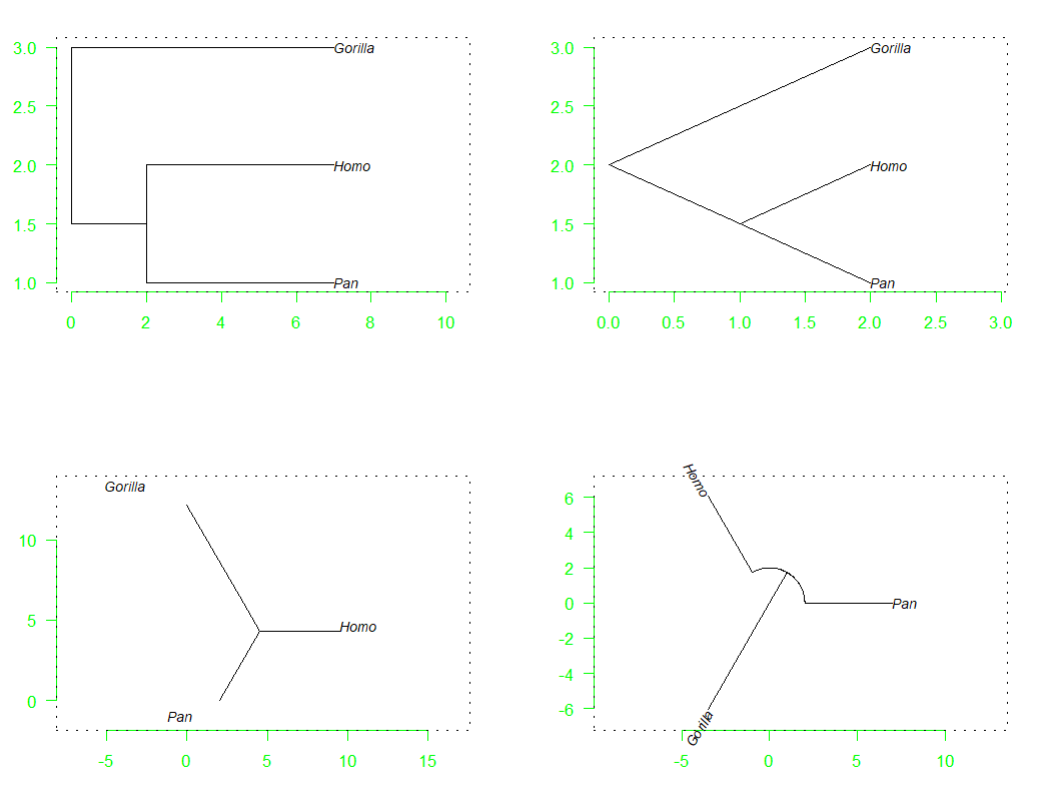
class(mytr) # 查看“mytr”对象的类型。
args(plot.phylo) # 查看plot.phylo相关参数
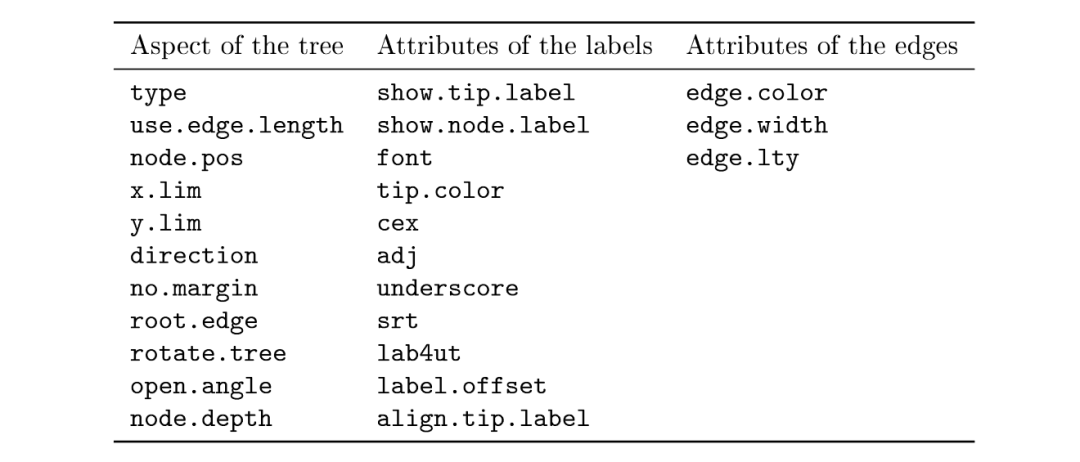
geo <- factor(c("Africa", "World", "Africa")) # 把地理分布赋值到变量mycol <- c("blue", "red")[geo] # 根据geo变量添加颜色plot(mytr, tip.color = mycol) # 画出指定颜色的系统发育树
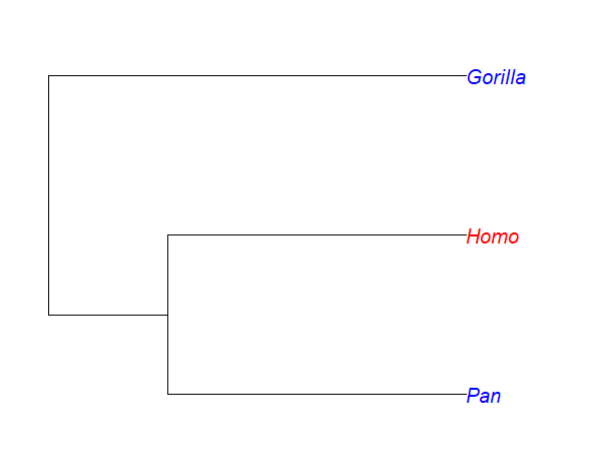
par(xpd = TRUE)plot(mytr, tip.color = mycol, cex = c(1, 1, 1.5))
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果
往期回顾
|
01 |
|
02 |
|
03 |
|
04 |