一文搞定生信分析强助攻-药敏分析(pRRophetic和oncoPredict包)

if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install(c("limma", "car", "ridge", "preprocessCore", "genefilter", "sva"))使用 "BiocManager" 包来安装多个其他包,包括 "limma"、"car"、"ridge"、"preprocessCore"、"genefilter" 和 "sva"。install.packages("ggplot2")install.packages("ggpubr")引用包library(limma)library(ggpubr)library(pRRophetic)
library(ggplot2)#加载所需的R包,包括 "limma"、"ggpubr"、"pRRophetic"、"ggplot2" 等set.seed(12345)pFilter=0.001 #pvalue的过滤条件设置显著性过滤条件,用于筛选差异显著的结果gene="VCAN" #目标基因expFile="symbol.txt" #表达数据文件setwd("") #设置工作目录#获取药物列表data(cgp2016ExprRma)#载入已经提供的 "cgp2016ExprRma" 数据集data(PANCANCER_IC_Tue_Aug_9_15_28_57_2016)allDrugs=unique(drugData2016$Drug.name)#载入已经提供的 "PANCANCER_IC_Tue_Aug_9_15_28_57_2016" 数据集。#读取表达输入文件,并对数据进行处理rt=read.table(expFile, header=T, sep="t", check.names=F)

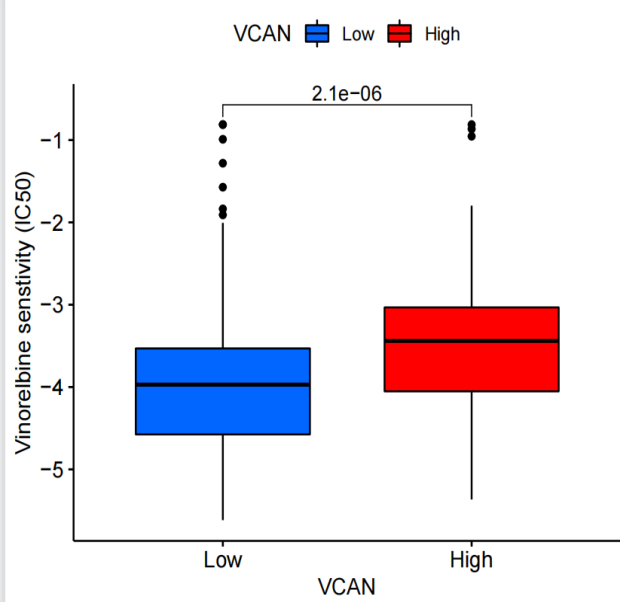
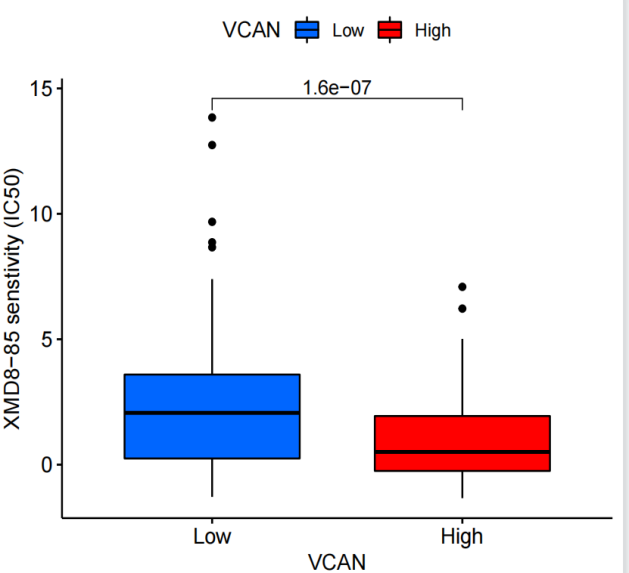
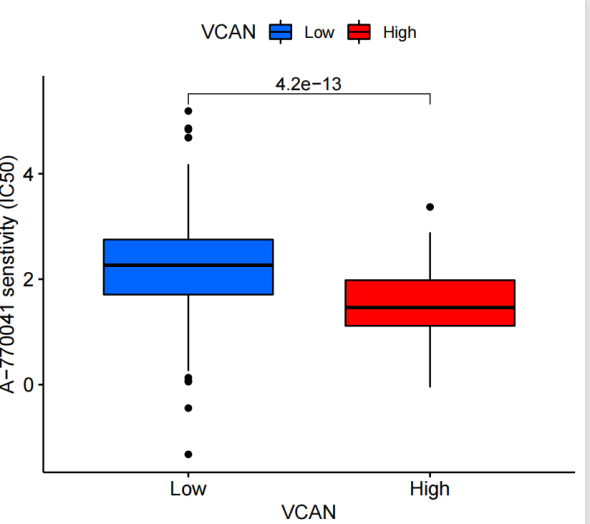
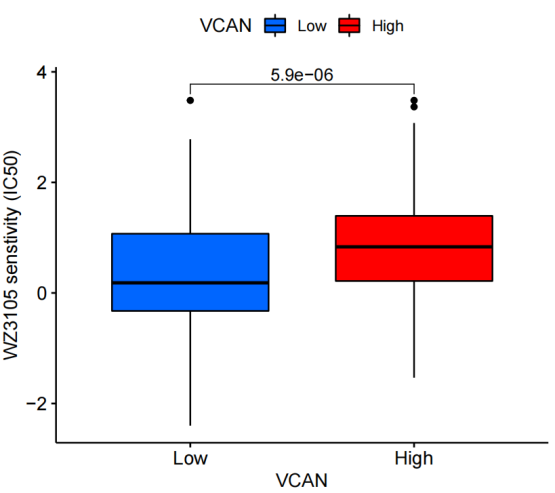
rt=as.matrix(rt)rownames(rt)=rt[,1]exp=rt[,2:ncol(rt)]# 将处理后的矩阵中的表达数据提取出来。dimnames=list(rownames(exp),colnames(exp))#创建矩阵 data 设置维度名称。#在R中,维度名称允许你为矩阵的行和列指定标签,以便更好地理解矩阵中的数据data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)#将数据矩阵 exp 转换为 data 矩阵。具体步骤如下:# as.matrix(exp): 将数据框 exp 转换为数值型矩阵。# as.numeric(...): 将数值型矩阵中的数据转换为数值。# matrix(...): 使用转换后的数值和指定的行数(由 nrow(exp) 给出)和维度名称(由 dimnames 给出)创建一个新的矩阵 data。#删掉正常样品data=avereps(data)#函数 avereps 对矩阵 data 进行处理。avereps 函数的目的是求每一行的平均值data=data[rowMeans(data)>0.5,]#将矩阵 data 进一步处理,只保留行平均值大于0.5的行。#排除那些平均表达量较低的样本,以便更关注表达丰度较高的样本。group=sapply(strsplit(colnames(data),"\-"), "[", 4)#将矩阵 data 的列名进行处理,提取出每列名称的第四个部分(使用 "-" 作为分隔符)。用于从样本名中获取样本的分组信息。group=sapply(strsplit(group,""), "[", 1)#将每个字符串中的每个字符作为单独的元素。然后,使用 [ 函数提取每个字符串的第一个字符。这可能是为了进一步处理样本分组信息。group=gsub("2","1",group)#用 gsub 函数将所有字符串中的字符 "2" 替换为 "1"。这可能是将样本分组信息进行标准化或重新编码。data=data[,group==0]#将矩阵 data 进一步处理,只保留样本分组信息为 "0" 的列。这可能是为了只保留特定样本分组的数据。data=t(data)#对矩阵 data 进行转置,将行列互换rownames(data)=gsub("(.*?)\-(.*?)\-(.*?)\-.*", "\1\-\2\-\3", rownames(data))#用正则表达式处理矩阵 data 的行名,从中提取出所需的信息,并重新设置行名。data=t(avereps(data))#根据目标基因的表达量对样品进行分组geneExp=as.data.frame(t(data[gene,,drop=F]))geneExp$Type=ifelse(geneExp[,gene]>median(geneExp[,gene]), "High", "Low")#从矩阵 data 中选择与目标基因相关的行,#并使用逗号 , 来指定列的选择。drop=F 参数确保即使只选择一列,结果也会保持为矩阵而不是向量。for(drug in allDrugs){#预测药物敏感性possibleError=tryCatch({senstivity=pRRopheticPredict(data, drug, selection=1, dataset = "cgp2016")},error=function(e) e)#使用 tryCatch 函数,尝试执行药物敏感性预测。#如果在执行过程中出现错误,将在错误处理函数中捕获错误并存储在 possibleError 中。if(inherits(possibleError, "error")){next}#如果 possibleError 包含了一个错误,那么这行代码将跳过当前药物,继续下一个循环迭代。senstivity=senstivity[senstivity!="NaN"]#移除药物敏感性结果中的 "NaN" 值,将有效的药物敏感性值存储在变量 senstivity 中。senstivity[senstivity>quantile(senstivity,0.99)]=quantile(senstivity,0.99)#对药物敏感性值进行处理,将高于 0.99 分位数的值设置为 0.99。这可能是为了去除极端异常值,以获得更好的可视化效果#将表达数据与药物敏感性的结果进行合并sameSample=intersect(row.names(geneExp), names(senstivity))#找到 geneExp 中与药物敏感性结果具有相同样本名称的样本。geneExp=geneExp[sameSample, "Type",drop=F]#从 geneExp 中选择与药物敏感性结果相同的样本,仅保留 "Type" 列。senstivity=senstivity[sameSample]#从药物敏感性结果中选择与 geneExp 相同样本的敏感性值。rt=cbind(geneExp, senstivity)#将基因表达分组和药物敏感性结果合并到一个数据框中。#设置比较组rt$Type=factor(rt$Type, levels=c("Low", "High"))#将 "Type" 列转换为因子,设定水平为 "Low" 和 "High"。type=levels(factor(rt[,"Type"]))comp=combn(type, 2)my_comparisons=list()#创建一个空列表来存储分组比较for(i in 1:ncol(comp)){my_comparisons[[i]]<-comp[,i]}#将所有可能的分组比较添加到 my_comparisons 列表中。#获取高低表达组差异的pvaluetest=wilcox.test(senstivity~Type, data=rt)#执行 Wilcoxon 秩和检验,检验药物敏感性是否在不同基因表达分组之间存在显著差异。diffPvalue=test$p.valueif(diffPvalue <pFilter){< span></pFilter){<>#绘制箱线图boxplot=ggboxplot(rt, x="Type", y="senstivity", fill="Type",xlab=gene,ylab=paste0(drug, " senstivity (IC50)"),legend.title=gene,palette=c("#0066FF","#FF0000"))+stat_compare_means(comparisons=my_comparisons)pdf(file=paste0("durgSenstivity.", drug, ".pdf"), width=5, height=4.5)#创建一个 PDF 文件,用于保存绘制的箱线图,文件名根据当前药物命名。print(boxplot)dev.off()}}
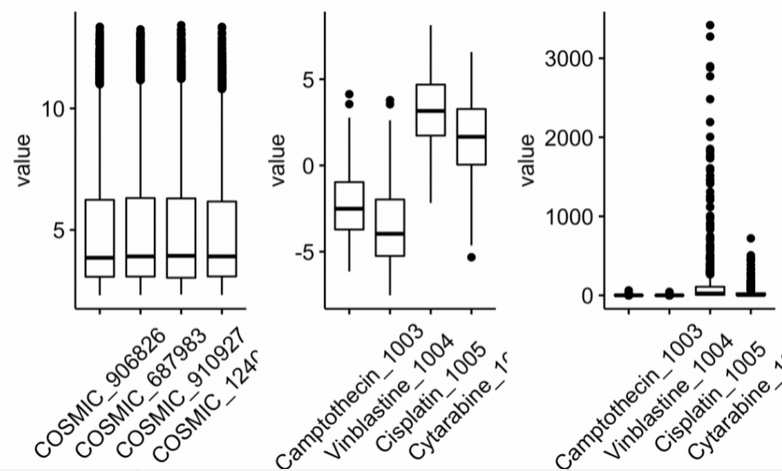
library(reshape2)library(ggpubr)th=theme(axis.text.x = element_text(angle = 45,vjust = 0.5))dir='./DataFiles/Training Data/'GDSC2_Expr = readRDS(file=file.path(dir,'GDSC2_Expr (RMA Normalized and Log Transformed).rds'))dim(GDSC2_Expr)GDSC2_Expr[1:4, 1:4]boxplot(GDSC2_Expr[,1:4])df=melt(GDSC2_Expr[,1:4])head(df)p1=ggboxplot(df, "Var2", "value") +th# Read GDSC2 response data. rownames() are samples, colnames() are drugs.dirGDSC2_Res = readRDS(file = file.path(dir,"GDSC2_Res.rds"))dim(GDSC2_Res) # 805 198GDSC2_Res[1:4, 1:4]p2=ggboxplot(melt(GDSC2_Res[ , 1:4]), "Var2", "value") +th ; p2# IMPORTANT note: here I do e^IC50 since the IC50s are actual ln values/log transformed already, and the calcPhenotype function Paul #has will do a power transformation (I assumed it would be better to not have both transformations)GDSC2_Res <- exp(GDSC2_Res)p3=ggboxplot(melt(GDSC2_Res[ , 1:4]), "Var2", "value") +th ; p3library(patchwork)p1+p2+p3
ggboxplot(melt(GDSC2_Res[ 1:4 ,]), "Var1", "value") +th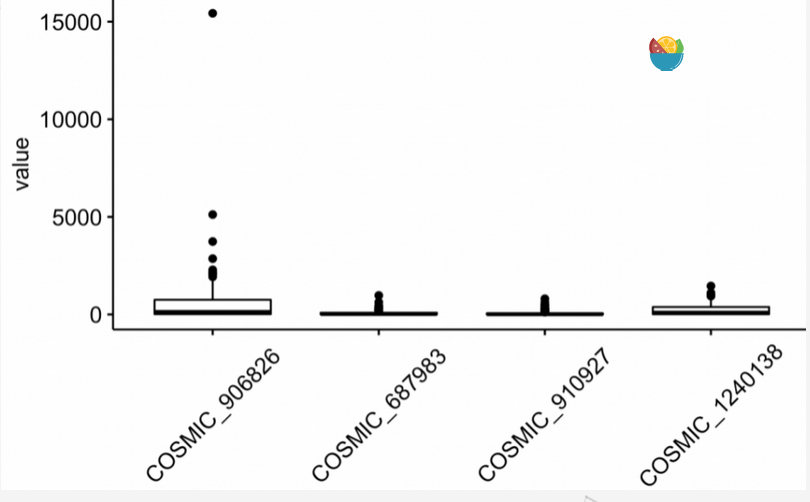
round(apply(GDSC2_Res[ 1:4 ,], 1, function(x){return(c(head(sort(x)),tail(sort(x))))}),2)
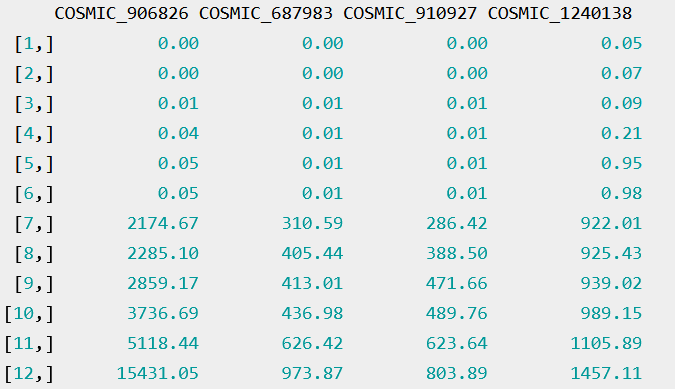
每个细胞系都是有自己的特异性药物和废物药物,IC50接近于0的就是神药 。表达量矩阵是从Genomics of Drug Sensitivity in Cancer (GDSC) 的
v2里面随机挑选10个细胞系作为要预测的矩阵。读入训练集的表达量矩阵 和药物处理信息。
install.packages("oncoPredict")rm(list = ls()) ## 魔幻操作,一键清空~options(stringsAsFactors = F)library(oncoPredict)library(data.table)library(gtools)library(reshape2)library(ggpubr)th=theme(axis.text.x = element_text(angle = 45,vjust = 0.5))dir='./DataFiles/Training Data/'GDSC2_Expr = readRDS(file=file.path(dir,'GDSC2_Expr (RMA Normalized and Log Transformed).rds'))GDSC2_Res = readRDS(file = file.path(dir,"GDSC2_Res.rds"))GDSC2_Res <- exp(GDSC2_Res)testExpr<- GDSC2_Expr[,sample(1:ncol(GDSC2_Expr),10)]testExpr[1:4,1:4]colnames(testExpr)=paste0('test',colnames(testExpr))dim(testExpr)calcPhenotype(trainingExprData = GDSC2_Expr,trainingPtype = GDSC2_Res,testExprData = testExpr,batchCorrect = 'eb', # "eb" for ComBatpowerTransformPhenotype = TRUE,removeLowVaryingGenes = 0.2,minNumSamples = 10,printOutput = TRUE,removeLowVaringGenesFrom = 'rawData' )#解读药物预测结果library(data.table)testPtype <- fread('./calcPhenotype_Output/DrugPredictions.csv', data.table = F)testPtype[1:4, 1:4]
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |