Cell Ranger轻松搞定单细胞上游分析
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

-
又一次,小果准时与大家相见啦。今天和小果一起来学习单细胞上游分析流程吧。
单细胞测序上游分析主要使用Cell Ranger软件。Cell Ranger 是一个用于单细胞基因表达分析的软件包,由 10x Genomics 公司开发。它包含五个与3’单细胞基因表达方案及相关产品相关的流程:cellranger mkfastq、cellranger count、cellranger aggr、cellranger reanalyze 和 cellranger vdj。其中,cellranger mkfastq 的作用是将Illumina测序仪生成的原始base call(BCL)文件解析成FASTQ格式的文件。它是Illumina的bcl2fastq的封装,并带有特定于10x Genomics文库的附加功能。
我们可以从10x Genomics的官方网站上下载和安装Cell Ranger软件,具体软件使用可以参考官网https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger。
整个单细胞上游流程和cell Ranger的使用如图所示。

小果是按照以下流程来操作的:
软件下载
mkdir yard #新建文件夹cd ../yard #进入此文件夹mkdir appscd apps#下载cellranger软件并解压curl -o cellranger-7.1.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-7.1.0.tar.gz?Expires=1680536714&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cHM6Ly9jZi4xMHhnZW5vbWljcy5jb20vcmVsZWFzZXMvY2VsbC1leHAvY2VsbHJhbmdlci03LjEuMC50YXIuZ3oiLCJDb25kaXRpb24iOnsiRGF0ZUxlc3NUaGFuIjp7IkFXUzpFcG9jaFRpbWUiOjE2ODA1MzY3MTR9fX1dfQ__&Signature=nrDGafilO0qCsE0KPOKhyFZPdj~go-zAZhACPT6v~Rjd9fPibaE28B22726ytq1WZNJfwBysfQMI3Xo3zEbjc8gJZv-141alIGts5eyXj-JpMgu6mxVPJQ3jKMopkP~B6vNyOR1R9h5IIccHZD~RlKL0wT-cXDgefvJoXoqmx0Lxt6Jpvuoekm0h2l~AKF~m4ARSItKFeQ-W2OxF2BOaMlyXIYPYOhcESApA9TabRqFUnhGUssld1nJdfSfGR-uFHH05AxUK6D1~LtTO5whUNUib8FHsYRwdzieFjy-Xc7i7KPcvPGwnUecn4RnC4gPBlJ3oBmEArTQedRE4rlTtWA__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"tar -zxvf cellranger-7.1.0.tar.gz #解压#为方便使用,将软件路径设置为变量PATHexport PATH=/mnt/home/user.name/yard/apps/cellranger-6.1.2:$PATH

准备fastq文件和参考基
1.下载参考基因组数据:
可直接下载官网提供的已构建好的索引文件,https://www.10xgenomics.com/resources/datasets/ ,建议下载最新版本并进行解压。
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz tar -zxvf refdata-gex-GRCh38-2020-A.tar.gz2.fastq文件
若是BCL原始的测序数据,须使用cellranger mkfastq将其转换为fastq格式文件。
cellranger mkfastq --id=tutorial_walk_through #输出文件夹--run=/mnt/home/user.name/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-1.2.0 #BCL文件文件夹--csv=/mnt/home/user.name/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-simple-1.2.0.c #csv文件
若是fastq文件,可直接run cellranger count进行定量,得到表达矩阵。如果有多个生物学样本或一个样本有多个重复/文库时,可以使用cellranger aggr进行整合。
fastq文件如下所示:
scdata_1k_v3_fastqs/ scdata_1k_v3_fastqs/scdata_1k_v3_S1_L001_R2_001.fastq.gz scdata_1k_v3_fastqs/scdata_1k_v3_S1_L002_I1_001.fastq.gz scdata_1k_v3_fastqs/scdata_1k_v3_S1_L001_R1_001.fastq.gz scdata_1k_v3_fastqs/scdata_1k_v3_S1_L002_R1_001.fastq.gz scdata_1k_v3_fastqs/scdata_1k_v3_S1_L002_R2_001.fastq.gz scdata_1k_v3_fastqs/scdata_1k_v3_S1_L001_I1_001.fastq.gz#I1 Index#R1 barcode+UMI#R2 reads

运行cellranger
1.cellranger-count
Count gene expression (targeted or whole-transcriptome) and/or feature barcode reads from a single sample and GEM wellcellranger count --id=run_count_1kscdatas #输出文件夹的名字 --fastqs=/mnt/home/user.name/yard/run_cellranger_count/scdata_1k_v3_fastqs #测序数据的路径--sample=scdata_1k_v3 #指定sample的配套文件前缀 --transcriptome=/mnt/home/user.name/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A #参考基因组路径cellrangercount.shcellranger=../cellranger-6.1.2/bin/cellrangerdb=../refdata-gex-GRCh38-2020-Afq_dir=../fastq/$cellranger count --id=test1 #输出文件夹的名字--fastqs=$fq_dir #测序数据的路径--sample=scdata_1k_v3 #指定sample的配套文件前缀--transcriptome=$db #参考基因组路径
2.cellranger aggr
①Create aggregation CSV
The CSV file is a two-column file. The first column is for the sample id. This id name can be anything you want. Choose descriptive ids since they are used later in the analysis. The second column contains the paths to themolecule_info.h5 output files from the cellranger count pipelines.
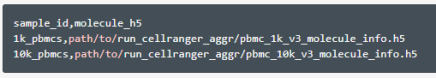
②运行代码
cellranger aggr --id=1k_10k_scdata_aggr--csv=scdata_aggr.csv
Cellranger的流程就如上所示,大家跟着小果一起实践一下吧。
我们对测序数据进行分析时,必定会用到比对软件或者工具,小果在这里给大家推荐一个在线小工具:fasta序列多重比对(http://www.biocloudservice.com/321/321.php),支持输入数据,快速得到结果。大家在之后比对时可以用起来哦。
好啦,不多说了,今天的内容就到这里了,你学会了吗!

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓