ggplot2 之作:多彩和谐的通路富集可视化之旅 !
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

hello,今天小果来教大家如何对KEGG富集分析的结果通过ggplot2实现可视化并绘制对应的KEGG通路网络图 ,让你的分析结果更加美观!感兴趣的话就和小果一起来看一下吧!
1. 准备需要的R包
先来简单介绍一下我们今天用到的R包,分别是DOSE、clusterProfiler、org.Hs.eg.db、ggplot2、tidyverse以及enrichplot包。同学们要提前下载好哦!在这里小果为大家简单的介绍一下其中几个R包:
1.DOSE:DOSE包主要用于基因功能富集分析,它能够通过比较输入的基因列表与已知的基因功能注释信息,找出在特定功能类别中富集的基因。
2.clusterProfiler:clusterProfiler是一个用于生物信息学分析的R包,主要用于功能注释和富集分析。它提供了针对多种生物数据类型的功能富集分析方法,包括GO、KEGG、Reactome等。该包还提供了可视化工具,使用户能够更好地理解和解释功能富集结果。
3.org.Hs.eg.db:org.Hs.eg.db是一个R语言中用于人类基因注释的数据库包。它提供了对人类基因组的注释信息,包括基因ID、基因名称、基因符号、通路信息等。使用org.Hs.eg.db包,用户可以将基因ID转换为易读的基因符号,从而更容易理解和解释功能富集结果。
4.tidyverse:tidyverse是一个由多个R包组成的集合,旨在提供一套一致且易于使用的数据处理和可视化工具。这些包包括ggplot2、dplyr、tidyr、readr等。其中,ggplot2是用于数据可视化的重要组成部分,提供了强大且灵活的绘图功能,使用户能够创建高质量的图表,用于数据探索和展示。
5.enrichplot:enrichplot是一个用于生物信息学分析的R包,主要用于功能富集结果的可视化。它提供了多种绘图函数,可以绘制基因集的富集分析结果,如GO条形图、KEGG通路网络图等。enrichplot提供了丰富的参数设置,以满足用户对结果可视化的个性化需求。
(关注小果回复111领取相关文件哟!)
2. KEGG富集分析
enrichKEGG进行富集分析
首先,加载DOSE包并使用该包中的基因集作为我们分析的对象:
library("DOSE")data(geneList, package = "DOSE")g_list <- names(geneList)[1:100]head(g_list)
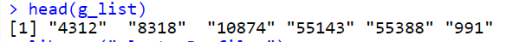
利用enrichKEGG函数进行富集分析,对指定的基因列表进行KEGG通路富集分析,并设置p-value和q-value的阈值。
library("clusterProfiler")library("org.Hs.eg.db")ek <- enrichKEGG(gene = g_list,organism = "hsa",pvalueCutoff = 0.05,qvalueCutoff = 0.05)write.table(ek, file = "ek.txt", sep = "t", quote = FALSE, row.names = FALSE)
注意:
·pvalueCutoff = 0.05:设置p-value的阈值,用于筛选富集分析结果中统计显著的通路。在这里,设定为0.05,即p-value小于0.05的通路被认为是显著富集的通路。
·qvalueCutoff = 0.05:设置q-value的阈值,q-value是经过FDR校正的p-value,用于控制多重假设检验的错误发现率。同样,设定为0.05,即q-value小于0.05的通路被认为是显著富集的通路。
至此我们已经完成了富集分析的流程,让我们一起来看一下分析的结果吧!
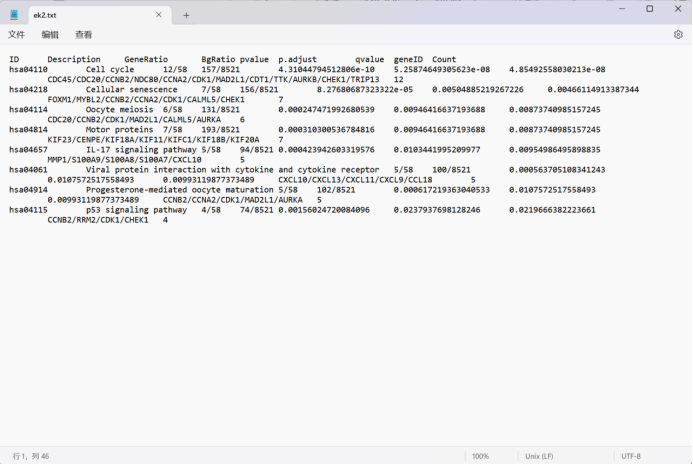
需要注意的是结果中geneID为EntrezID。
head(ek@result$geneID)
将EntrezID转换为Symbol
使用setReadable函数将EntrezID转换为Symbol:
ek2 <- setReadable(ek,OrgDb = "org.Hs.eg.db",keyType = "ENTREZID")

ggplot2可视化结果
首先,我们需要通过安装tidyverse包并读取保存的富集分析结果文本文件,对表格进行处理和计算Enrichment Factor,在这里小果用tidyverse包的separate函数将GeneRatio和BgRatio的分子分母先分开,再进行计算:
library(tidyverse)ek.rt <- read.table("ek.txt", header = TRUE, sep = "t", quote = "")ek.rt <- separate(data = ek.rt, col = GeneRatio, into = c("GR1", "GR2"), sep = "/")ek.rt <- separate(data = ek.rt, col = BgRatio, into = c("BR1", "BR2"), sep = "/")ek.rt <- mutate(ek.rt, enrichment_factor = (as.numeric(GR1) / as.numeric(GR2)) / (as.numeric(BR1) / as.numeric(BR2)))
筛选出前10个通路进行可视化:
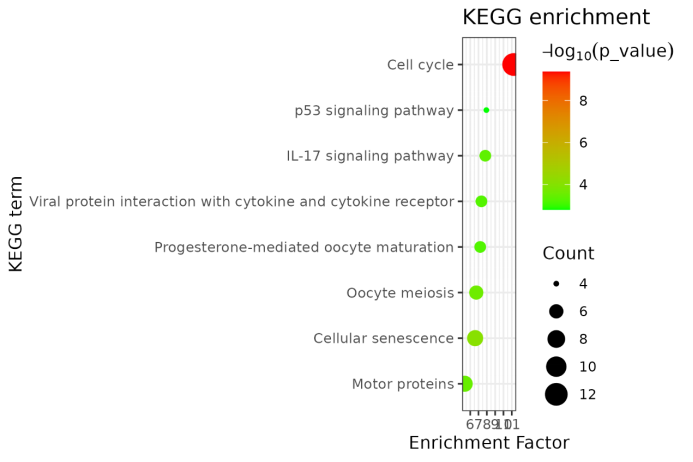
ek.rt10 <- ek.rt %>% filter(row_number() >= 1, row_number() <= 10)ggplot2绘制dotplot
使用ggplot2包绘制Enrichment Factor与KEGG term之间的关系图:
library("ggplot2")p <- ggplot(ek.rt10, aes(enrichment_factor, fct_reorder(factor(Description), enrichment_factor))) +geom_point(aes(size = Count, color = -1 * log10(pvalue))) +scale_color_gradient(low = "green", high = "red") +labs(color = expression(-log[10](p_value)), size = "Count",x = "Enrichment Factor", y = "KEGG term", title = "KEGG enrichment") +theme_bw()ggsave("er.rt10_KEGG.png", width = 6, height = 4)
我们一起来看看效果图吧!
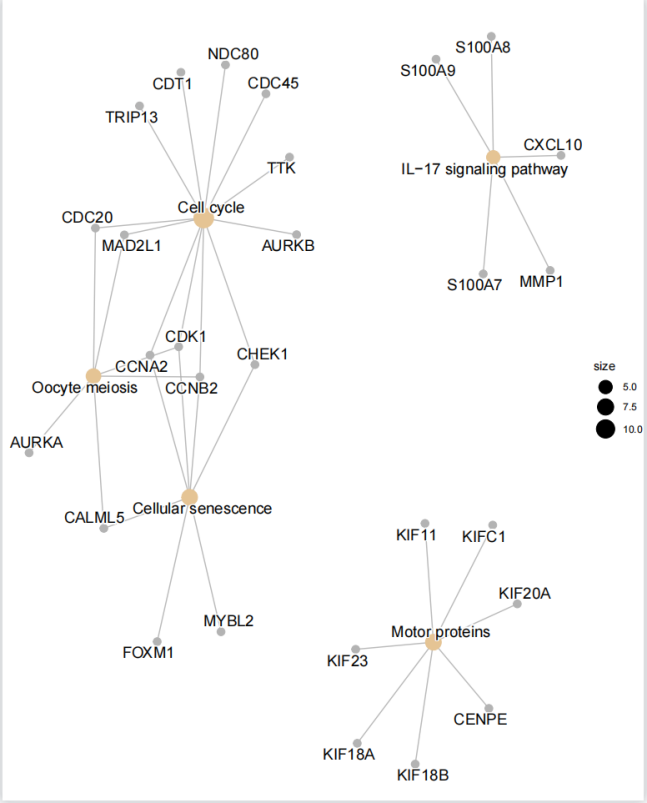
绘制KEGG通路网络图
使用enrichplot包的cnetplot函数绘制KEGG通路网络图,并将结果保存为PDF文件。
library("enrichplot")pdf(file = "net-pathway.pdf", width = 8, height = 10)cnetplot(ek2)dev.off()
效果图如下:
以上教程小果介绍了如何使用R语言中的DOSE和clusterProfiler包进行基因集的KEGG通路富集分析,并通过ggplot2和enrichplot实现可视化化。通过这些代码,你可以将自己的基因列表进行富集分析,并进一步将结果进行可视化以帮助解释和理解数据哦!怎么样,你学会了嘛?
机器学习相关其他分析内容欢迎尝试本公司新开发的云平台生物信息分析小工具,零代码完成分析,云平台网址:http://www.biocloudservice.com/home.html。
点击“阅读原文”立刻拥有
↓↓↓