生信大杀器之神奇的贝叶斯网络(6)

导入R包
1.# 加载所需的R包2.library(edgeR)3.library(bnlearn)加载数据现在我们开始加载需要的数据,通过read_excel()函数读取相应的xlsx文件1.# 加载数据2.# 使用readxl包中的read_excel函数读取Excel文件中的数据,文件路径需要根据你的实际情况修改。3.wulffenTable <- read_excel("C:/Users/17240/Desktop/GSE71562_E14R012_raw_counts.xlsx")4.# 显示数据的前几行5.head(wulffenTable)6.7.# 选择需要分析的数据,这里选择了第2到19列的数据8.X = wulffenTable[2:19]9.colnames(X)<-c("at0","at0.5","at1","at2","at5","at10","bt0","bt0.5","bt1","bt2","bt5","bt10","ct0","ct0.5","ct1","ct2","ct5","ct10")
/2_WMmqY8ELiakW7ANPDJJWs7wlWwpRyg.png)
根据先验知识预设贝叶斯网络结构
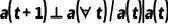 .根据这个条件,我们可以预设贝叶斯网络的结构:
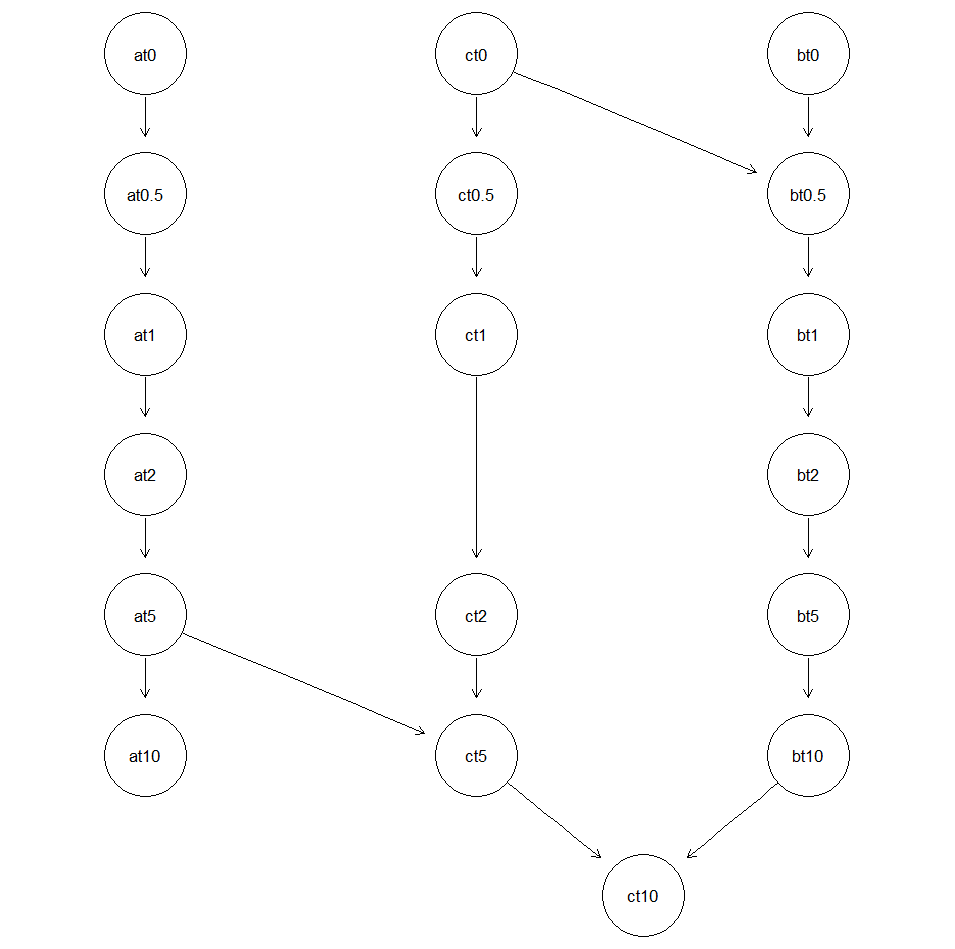
.根据这个条件,我们可以预设贝叶斯网络的结构:1.2.w=data.frame(from=c("at0","at0.5","at1","at2","at5","bt0","bt0.5","bt1","bt2","bt5","ct0","ct0.5","ct1","ct2","ct5"),3. to=c("at0.5","at1","at2","at5","at10","bt0.5","bt1","bt2","bt5","bt10","ct0.5","ct1","ct2","ct5","ct10"))
创建贝叶斯网络并调整
1.pdag = gs(X,whitelist = w)2.pdag3.pp = graphviz.plot(pdag)

1.pdag = gs(X,whitelist = w,blacklist = c("bt0.5","ct0"))2.pdag4.pp = graphviz.plot(pdag)
/5_2NIRHv1kiaeu566oFDH625dnnPQCLA.png)
1.pdag = set.arc(pdag, from = "at5", to = "ct5")2.pdag = set.arc(pdag, from = "bt10", to = "ct10")3.pp = graphviz.plot(pdag)
参数学习
1.#拆分数据为训练数据集和测试数据集2.wul.train=X[1:4000,]3.wul.test=X[4001:4319,]4.set.seed(123)5.fitted = bn.fit(pdag, wul.train)6.fitted
预测
1.pred = predict(fitted, node = "ct5", data = wul.test, method = "bayes-lw")2.cbind(wul.test$ct5,pred)3.plot(wul.test$ct5-pred)
/7_OUa545psnW5lfdrnz9a1xh6FmQ3w2w.png)
/8_TAtNFMxE1hEmxtdmUKzl41icSxribA.png)
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!
/9_B2jkicacLzPjw4xMSqVvoucZjonVDA.jpg)
定制生信分析
服务器租赁
扫码咨询小果

/11_9N02aBaau07RufXeXRLmvAHsrFZxtw.jpg)
往期回顾
|
01 |
|
02 |
|
03 |
|
04 |