3种机器学习方法同时结合预后生存期展开分析!确定不进来看看?(一)
点击蓝字 关注我们
小伙伴们大家好啊,今天大海哥决定要给大家准备一下非常厉害的教学,3种机器学习算法同时结合预后生存期分析!通过代码实践的方式让你彻底学会如何将机器学习与生物信息学完美融合,看到这很多小伙伴肯定等不及了,心急吃不了热豆腐,让我们先来了解一下今天的分析案例的背景,然后再来深入学习~让我们开始吧!
今天我们的分析案例是一个乳腺癌生存率的预测案例,我们知道癌症患者临床决策中最重要的部分是准确估计预后和生存时间。而同一疾病阶段和相同临床特征的乳腺癌患者可能有不同的治疗反应和总生存期,因此,提供一种简单的算法来预测乳腺癌患者的生存可能对临床决策过程有很大帮助。为此,随机森林和支持向量机(SVM)是本次分析中使用的两种监督学习方法。为什么是2种,不是3种吗?是的,因为针对SVM算法大海哥使用了多项式核SVM和径向内核SVM两种,所以一共是3种!
了解完背景知识,就让我们进入代码的世界吧!
#加载本次分析需要的R包library(corrplot)library(VIM)library(ggplot2)library(gridExtra)library(scales)library(tidyr)library(dplyr)library(readr)library(ggformula)library(imputeMissings)library(gapminder)library(caret)library(rpart)library(rattle)library(kernlab)library(randomForest)library(DMwR2)library(car)library(MLeval)#R包有点多,因为一个完整的分析除了主要分析流程外,还需要一些探索性分析来对数据做一个初步的探索!#然后就是加载数据df <- read.csv("RNA.csv", header=TRUE, sep=",", stringsAsFactors = FALSE)dim(df)
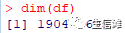
#这个数据集来自乳腺癌分子分类学国际联盟 (METABRIC)。该数据集包含 1904 名乳腺癌患者的 31 个临床属性、331 个基因的 mRNA 水平 z -score以及 175 个基因的突变#在这个数据中,“tumor_other_histologic_subtype”、“oncotree_code”、“cancer_type”和“cancer_type_detailed”表示不同的癌症类型名称。我们只用选一个就好,并且还有一个冗余的字段,我们都需要稍后把它们剔除掉。#用代码去除一下,并且看一下缺失数据df[df == ""] <- NAdf_t = df[, c(1:31)]#计算每个变量缺失数据的百分比M_Val <- function(x){mm_v=round((sum(is.na(x))/length(x))*100,3)}nam_1<-colnames(df_t)c<-apply(df_t,2,M_Val)df_p <- data.frame(nam_1,c)rownames(df_p)<-NULLdf_p1 <- df_p %>% filter(c > 5 ) %>% mutate(c1=ifelse(c < 5, c-.4, c-2.1))gf_boxplot(c~nam_1 , data = df_p) %>%gf_labs(title = "Missing data percentage",x = "",y = "Percent (%)")%>% gf_theme(axis.text.x=element_text(angle=65, hjust=1)) %>%gf_label(c1~nam_1,label =~ c,data = df_p1, col = "black",fill =~ nam_1, alpha = .2,show.legend = FALSE)
/2_lKH0u6O5YPTfrJrLOUCHo5nkghL0aQ.png)
#可以看到有几列数据的缺失值还挺多的#删除冗余列df$patient_id <- NULLdf$cancer_type <- NULLdf$death_from_cancer <- NULLdf$cancer_type_detailed <- NULLdf$oncotree_code <- NULLdf$cohort <- NULLdf$tumor_stage <- NULLdf$overall_survival_months <- NULLdf$X3.gene_classifier_subtype <- NULLdf$er_status_measured_by_ihc <- NULLdf$her2_status_measured_by_snp6 <- NULLclinical = df[, c(1:20)] #拿到全新的临床数据了head(clinical,2)#大概看一下数据

#删除具有一个属性的因子数据clinical <- clinical[!( clinical$tumor_other_histologic_subtype=="Metaplastic"| clinical$pam50_._claudin.low_subtype=="NC"),]clinical$tumor_other_histologic_subtype <- as.factor(clinical$tumor_other_histologic_subtype);clinical$pam50_._claudin.low_subtype <- as.factor(clinical$pam50_._claudin.low_subtype);#然后把一些关键变量转为因子clinical$type_of_breast_surgery <- as.factor(clinical$type_of_breast_surgery)clinical$cellularity <- as.factor(clinical$cellularity);clinical$chemotherapy <- as.factor(clinical$chemotherapy)clinical$pam50_._claudin.low_subtype <- as.factor(clinical$pam50_._claudin.low_subtype);clinical$er_status <- as.factor(clinical$er_status);clinical$neoplasm_histologic_grade <- as.factor(clinical$neoplasm_histologic_grade);clinical$her2_status <- as.factor(clinical$her2_status);clinical$tumor_other_histologic_subtype <- as.factor(clinical$tumor_other_histologic_subtype)clinical$hormone_therapy <- as.factor(clinical$hormone_therapy);clinical$inferred_menopausal_state<- as.factor(clinical$inferred_menopausal_state)clinical$integrative_cluster<- as.factor(clinical$integrative_cluster );clinical$primary_tumor_laterality<- as.factor(clinical$primary_tumor_laterality)clinical$pr_status<- as.factor(clinical$pr_status);clinical$radio_therapy <- as.factor(clinical$radio_therapy)clinical <- clinical %>% mutate(lymph_positive = ifelse(lymph_nodes_examined_positive == 0, "0", "1"))clinical$lymph_positive <- as.factor(clinical$lymph_positive); clinical$lymph_nodes_examined_positive <- NULLclinical$overall_survival<- as.factor(clinical$overall_survival);#然后删除缺失值clinical <- na.omit(clinical)#上面的那么多代码,都是为了我们后面的分析,知道数据预处理有多辛苦了吧!#然后就可以开始我们的探索性分析(EDA)啦!#先来看看变量之间的相关性吧!clinical_n <- select_if(clinical, is.numeric)corrmatrix <- cor(clinical_n)corrplot::corrplot(corrmatrix, method="shade", type = "upper",tl.cex=.6, tl.col="black", title="Correlation Plot",number.font = 2, mar=c(0,0,1,0), )
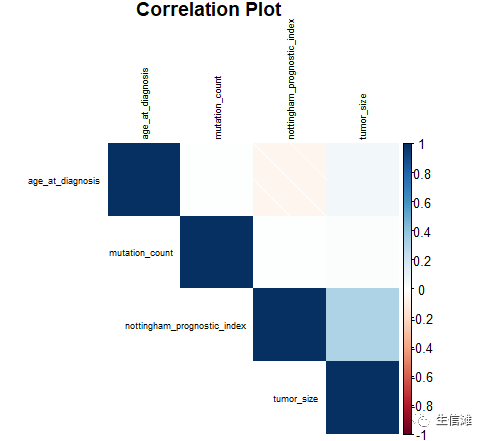
#好像关联不大
#那我们再来看看一些连续变量伴随生存结果的箱型图
#看看年龄的分布情况
/5_N7MwvB0xMTeibLT7Y4eWxJCHj7VHVQ.png)
#再看看突变的分布clinical %>% gf_boxplot(mutation_count~overall_survival) %>%gf_labs(x = "Survival",y = "Mutation") %>%gf_theme(axis.text = element_text(size = 12),axis.title = element_text(size = 12),legend.title=element_text(size=14),legend.text=element_text(size=9))
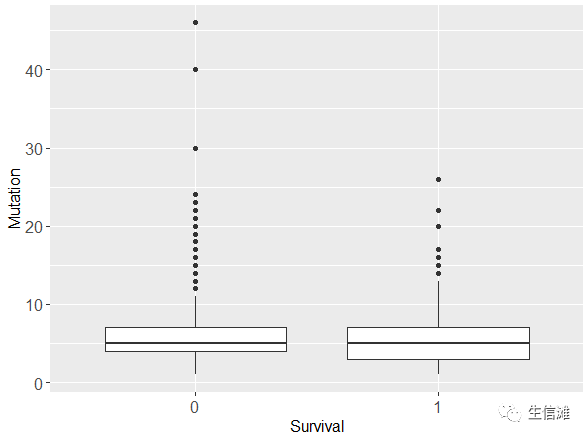
#最后看看肿瘤尺寸的分布clinical %>% gf_boxplot(tumor_size~overall_survival) %>%gf_labs(x = "Survival",y = "Tumor size") %>%gf_theme(axis.text = element_text(size = 12),axis.title = element_text(size = 12),legend.title=element_text(size=14),legend.text=element_text(size=9))
#可以看到肿瘤尺寸和突变的数据存在很多极大值,所以我们需要对肿瘤尺寸和突变处理一下clinical$tumor_size_T <- log10((clinical$tumor_size)); clinical$tumor_size <- NULLclinical$mutation_count_T <- log10((clinical$mutation_count)); clinical$mutation_count <- NULL#来看一下处理前后的数据维度par(mfrow=c(2 ,2))hist(clinical_n$tumor_size , main = "", xlab = "Tumor size", freq = FALSE )curve(dnorm(x, mean = mean(clinical_n$tumor_size), sd = sd(clinical_n$tumor_size)),col = "red", add = TRUE)hist(clinical$tumor_size_T , main = "", xlab = "Tumor size ", freq = FALSE )curve(dnorm(x, mean = mean(clinical$tumor_size_T), sd = sd(clinical$tumor_size_T)),col = "red", add = TRUE)hist(clinical_n$mutation_count, main = "", xlab = "Mutation count", freq = FALSE )curve(dnorm(x, mean = mean(clinical_n$mutation_count), sd = sd(clinical_n$mutation_count)),col = "red", add = TRUE)hist(clinical$mutation_count_T, main = "", xlab = "Mutation count", freq = FALSE )curve(dnorm(x, mean = mean(clinical$mutation_count_T), sd = sd(clinical$mutation_count_T)),col = "red", add = TRUE)
/8_5H1MHYuOpd0FpKGCYpqOP8709vbX2g.png)
可以看到处理之后,数据整体符合正态分布,就可以继续后续的分析啦!
但是今天的分析量已经很大了,所以大海哥决定在下一期文章中,再继续后续的分析,大家记得到时候继续来学习哦!本次的数据处理大家也要好好学习,最好亲自动手试一试哦!
(最后推荐一下大海哥新开发的零代码云生信分析工具平台包含超多零代码小工具,上传数据一键出图,感兴趣的小伙伴欢迎来参观哟。
网址:
http://www.biocloudservice.com/home.html)

生信滩公众号
/10_ocafb5YBQcZWdQRrtP2Xo7vaFWaecA.jpg)
/11_07T3ukOr5mxE6sbTMZvYNklUgT3oKg.jpg)
点击“阅读原文”进入网址