掌握CMplot:使用R轻松创建精美SNP密度图
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

-
你知道怎么可视化基因组的染色体上基因的密度分布状况吗?这个图要求你正确展示染色体上基因位点密度的分布信息,今天小果就来给大家科普一个简单又好用的R包,让你轻松绘制美观的SNP密度图,那么就和小果一起来看一下吧!
本教程将介绍如何准备数据、安装CMplot包、创建染色体地图以及如何自定义可视化参数。
准备数据
在使用CMplot包之前,需要准备好染色体数据。数据应包括以下散列信息:
· SNP位点名称(可自定义);
· 染色体号/染色体名称:这个指的是SNP位点对应的染色体是哪一条哦;
· SNP位置信息:每个SNP位点在染色体上的位置,通常使用基因组坐标表示。
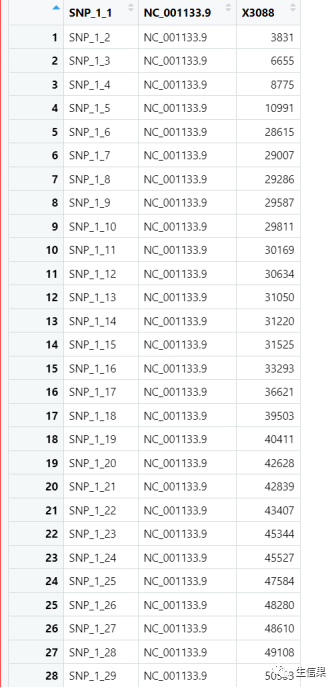
这些数据通常以文本文件的形式存储。文件应包含标题行,列标题应标识位置和值数据列,以便CMplot包正确解释数据。接下来小果给大家展示一下准备好的数据长什么样吧!

安装CMplot包
在开始使用CMplot包之前,需要先安装该包。您可以通过运行以下命令来安装CMplot包:
install.packages("CMplot")请注意,您需要确保您的计算机已连接到Internet,以便从CRAN下载和安装该包哦!

创建染色体SNP密度图
在安装了CMplot包之后,就可以使用该包中的CMplot()函数创建染色体地图。下面是一个示例代码:
library(CMplot) #导入CMplot包mydata <- read.table("SNP-new-old.txt", header = TRUE, sep = "t")CMplot(mydata, plot.type = "d", bin.size = 1e3, col = c("darkgreen", "yellow", "red"))在此示例中,我们使用read.table()函数读取名为“SNP-new-old.txt”的数据文件。该文件包含染色体上每个数据点的位置和值信息。我们将读取的数据存储在mydata变量中。
接下来,我们调用CMplot()函数。
plot.type参数设置为”d”,表示我们要绘制SNP密度图。小果科普:如果想要绘制堆积图,则可以将其设置为”stack”哦,如果想要绘制平滑的曲线图,则可以将其设置为”spline”。
bin.size参数设置为1,000,表示您想要将数据分成1,000个区间进行可视化。如果你的数据集很小,则可以将其设置为更小的值哦。反之如果你的的数据集很大,则可以将其设置为更大的值呢。
col参数指定了三种颜色,用于表示低密度、中等密度和高密度区域的数据。如果您想要使用不同的颜色,请将其替换为您选择的颜色。
好啦,到此为止你已经学会了怎么绘制SNP密度图,下面我们一起来看看效果吧!

更多生信绘图应用学习资源请大家移步小果专属云生信平台搜索更多资源哦!

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓
