R语言包compiler带你解锁生物学数据分析新境界
R语言自诞生以来,凭借其强大的数据处理和统计分析能力,迅速成为生物学家们的首选工具。其丰富的拓展包使得R能够应对各类数据处理任务,从基本的数据操作到高级的统计建模,皆能得心应手。然而,随着数据规模的不断扩大,R的解释执行模式在处理大数据时表现出了一定的性能瓶颈,因此生物学家们寻求着更高效的数据处理方式。
在R的海量拓展包中,compiler包脱颖而出,为研究人员提供了编译技术的威力,极大地提升了生物学数据分析的速度和性能。本文将介绍R语言包compiler,探索其在生物学数据分析领域的重要作用。
R语言包compiler是一款功能强大且广受欢迎的包,其主要作用是将R代码转换为二进制代码,从而提高代码执行效率,优化计算性能。在生物学领域,许多数据处理和统计分析任务需要处理大规模数据集,因此使用compiler包可以显著加速这些任务的执行,为生物学研究带来更高的效率和准确性。
编译技术是计算机科学的一个重要分支,通过将高级源代码转换为底层机器代码,以提高程序的执行速度和效率。这种技术在计算密集型任务中表现出色,正是生物学数据分析所需要的。R语言包compiler便提供了这样的崭新视角,将编译技术引入到生物学数据分析中,极大地提高了执行效率。
compiler包为R语言提供了即时编译(Just-In-Time Compilation,JIT)功能,它能够将R代码转换为底层的二进制代码,并在运行时动态执行。这种编译方式消除了传统解释执行的瓶颈,从而大幅提高了代码的运行速度。在生物学研究中,许多复杂的数据处理任务,如基因表达分析、序列比对、蛋白质结构预测等,都需要处理海量的数据。通过使用compiler包,研究人员可以加速这些任务的执行,从而缩短分析时间,更快地获得研究结果。
要使用compiler包,可以在R中使用以下命令进行安装和加载:
install.packages("compiler") #安装compiler语言包> library(compiler) #加载语言包
在生物学研究中,数据的可视化是非常重要的环节。R语言提供了许多用于数据可视化的包,如ggplot2、plotly等。然而,对于大规模数据集的可视化,仍然可能面临较长的绘图时间。通过使用compiler包,研究人员可以加速绘图函数的执行,实现更快速的数据可视化。这对于探索大规模生物学数据、发现模式和趋势,以及有效地传达研究结果至关重要。
示例:
假设我们有一个基因表达数据集,其中包含不同基因在多个样本中的表达量。我们将使用R语言的compiler包来进行数据处理和分析,并进行可视化展示。假设我们已经导入了以下示例数据:
# 假设我们已经导入了以下数据> gene_expression_data <- data.frame(+ Sample = c("Sample1", "Sample2", "Sample3", "Sample4", "Sample5"),+ Gene1 = c(10.2, 8.5, 12.1, 9.8, 11.3),+ Gene2 = c(5.6, 6.8, 4.2, 7.3, 5.1),+ Gene3 = c(2.3, 3.1, 2.8, 2.0, 3.5)+ )#安装并加载语言包> install.packages("compiler")> library(compiler)

我们可以使用compiler包来优化绘制基因表达量分布图的代码,以加快绘图过程。
# 创建一个函数来绘制基因表达量分布图> plot_gene_expression <- cmpfun(function(data, gene_name) {+ library(ggplot2)++ p <- ggplot(data, aes(x = Sample, y = !!sym(gene_name))) ++ geom_bar(stat = "identity", fill = "steelblue") ++ labs(title = paste("Gene Expression of", gene_name),+ x = "Sample",+ y = "Expression Level")++ print(p)+ })# 绘制基因1的表达量分布图> plot_gene_expression(gene_expression_data, "Gene1")
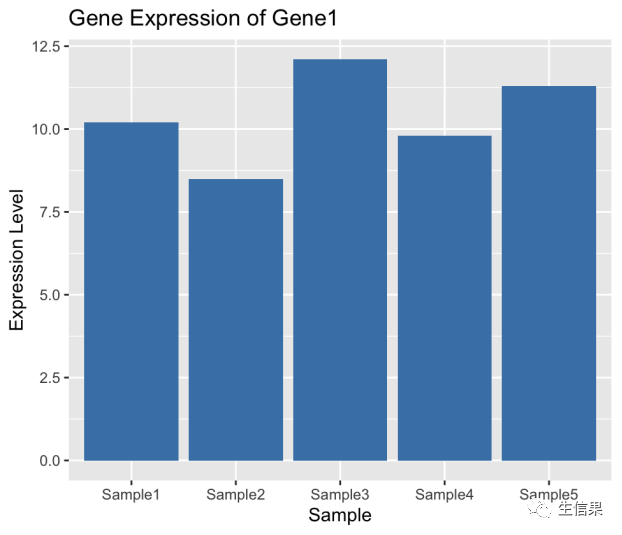
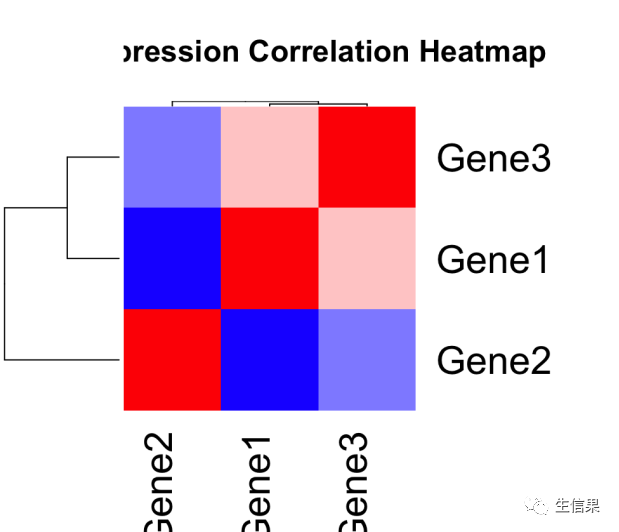
我们可以使用compiler包来加速计算基因表达的相关性,并绘制相关性热图。
# 创建一个函数来计算基因表达的相关性矩阵> calculate_correlation <- cmpfun(function(data) {+ cor_matrix <- cor(data[, -1], method = "pearson")+ return(cor_matrix)+ })# 计算基因表达的相关性矩阵> correlation_matrix <- calculate_correlation(gene_expression_data)# 绘制相关性热图> library(gplots)> heatmap.2(correlation_matrix,+ col = colorRampPalette(c("blue", "white", "red"))(100),+ key = TRUE,+ keysize = 1.0,+ symkey = FALSE,+ density.info = "none",+ trace = "none",+ margins = c(6, 10),+ main = "Gene Expression Correlation Heatmap")
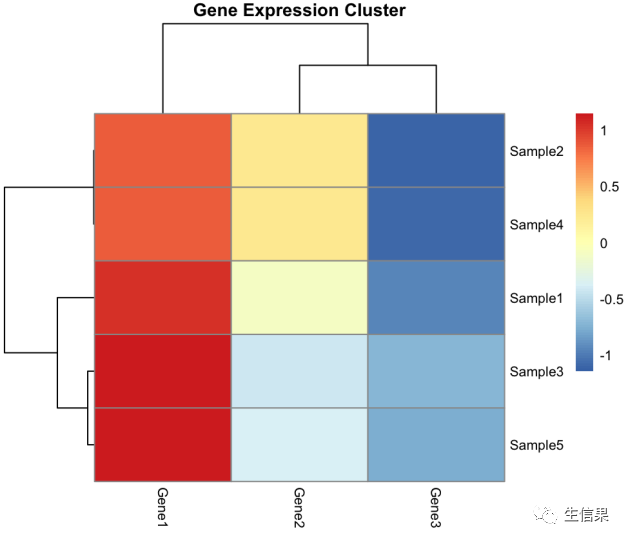
我们可以使用compiler包来加速基因表达聚类图的绘制过程。
# 创建一个函数来绘制基因表达聚类图> plot_gene_expression_cluster <- cmpfun(function(data) {+ library(pheatmap)++ rownames(data) <- data$Sample+ data <- data[, -1]++ pheatmap(data,+ clustering_method = "ward.D2",+ scale = "row",+ main = "Gene Expression Cluster",+ fontsize = 8)+ })# 绘制基因表达聚类图> plot_gene_expression_cluster(gene_expression_data)
以上示例演示了如何在R中使用compiler包对基因表达数据进行处理和分析,并绘制几个常见的生物学可视化图表。通过使用compiler包,我们可以显著加快数据处理和图表绘制的速度,提高生物学研究的效率和准确性。
一个不可忽视的优势是,compiler包的使用非常简单。只需在R代码中调用compiler函数,即可启用即时编译功能。此外,compiler包兼容大多数R语言的代码和函数,因此生物学家可以在现有的R脚本中轻松地应用compiler包,无需进行大规模的修改。
以上就是对R语言包compiler的简单介绍啦,它通过即时编译技术将R代码转换为高效的二进制代码,大幅提高了代码的执行效率,为生物学家处理大规模数据和进行复杂统计分析提供了强大支持。通过使用compiler包,生物学家可以更加高效地开展研究工作,加速数据处理和统计分析过程,从而为生物学研究的进展做出更大的贡献。
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小果哦~~
或者也可以关注我们的官网也会持续更新的哦~
往期推荐

