师妹带你分析单基因与肿瘤微环境评分的相关性—小提琴图
{ 点击蓝字,关注我们 }
我们对进行肿瘤微环境(TME)评分数据与基因表达数据的关联分析,并通过绘制小提琴图来展示不同肿瘤微环境得分组之间的差异,对分组之间基因表达数据的差异,进而了解肿瘤微环境得分在不同基因表达模式中的分布情况。
代码中对选定的基因vcan的样本表达量进行分析。下面是下载的肿瘤微环境评分分数文件,接下来绘制小提琴图。
#if (!requireNamespace("BiocManager", quietly = TRUE))# install.packages("BiocManager")#BiocManager::install("limma")#install.packages("reshape2")#install.packages("ggpubr")#检查是否已安装BiocManager包(用于安装Bioconductor中的包),#如果没有安装,则使用install.packages函数安装BiocManager包。#然后使用BiocManager::install函数安装limma包,该包用于后续的数据分析。#引用包library(limma)library(reshape2)library(ggpubr)#这两个包分别是reshape2和ggpubr,用于数据重塑和图形绘制expFile="geneExp.txt" #表达数据文件scoreFile="TMEscores.txt" #肿瘤微环境的打分文件#指定了表达数据文件(expFile)和肿瘤微环境打分文件(scoreFile)#读取表达数据文件读取表达数据文件(expFile)并进行预处理。#数据文件中的表达量数据被存储在名为exp的数据框中。exp=read.table(expFile, header=T, sep="t", check.names=F, row.names=1)gene=colnames(exp)[1]exp=exp[exp$Type=="Tumor",1,drop=F]exp$Type=ifelse(exp[,gene]>median(exp[,gene]), "High", "Low")#首先,通过colnames(exp)[1]取得第一列的列名(基因名),然后根据"Type"列筛选出"Tumor"类型的样本。#接着,根据基因表达量的中位数,将样本分为"High"和"Low"两组,并将结果存储在名为"Type"的列中。score=read.table(scoreFile, header=T, sep="t", check.names=F, row.names=1)score=score[,1:3]#读取肿瘤微环境打分文件,并对数据进行整理读取肿瘤微环境打分文件(scoreFile)并进行预处理。#数据文件中的肿瘤微环境打分数据被存储在名为score的数据框中。在这里,只保留前3列的数据,即包含肿瘤微环境得分的列。sameSample=intersect(row.names(exp), row.names(score))exp=exp[sameSample,"Type",drop=F]score=score[sameSample,,drop=F]rt=cbind(score, exp)rt$Type=factor(rt$Type, levels=c("Low", "High"))#样品取交集表达数据(exp)和肿瘤微环境打分数据(score)的样本取交集,以确保两者具有相同的样本。#然后将两个数据框按照样本进行合并,合并后的数据存储在名为rt的数据框中。#最后,将"Type"列转换为因子变量,并指定水平顺序为"Low"和"High"。data=melt(rt, id.vars=c("Type"))colnames(data)=c("Type", "scoreType", "Score")#将合并后的数据转换为ggplot2的输入文件使用melt函数将合并后的数据(rt)转换为ggplot2绘图函数的输入格式。#melt函数将数据框从宽格式转换为长格式,其中"Type"列作为id变量,其余的列被转换为两列:scoreType和Score。#绘制小提琴图p=ggviolin(data, x="scoreType", y="Score", fill = "Type",xlab="",ylab="TME score",legend.title=gene,add = "boxplot", add.params = list(color="white"),palette = c("blue","red"), width=1)p=p+rotate_x_text(45)p1=p+stat_compare_means(aes(group=Type),method="wilcox.test",symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1), symbols = c("***", "**", "*", " ")),label = "p.signif")#用ggviolin函数绘制小提琴图。小提琴图是一种可视化工具,用于显示不同组之间数值数据的分布情况。#x轴为scoreType,y轴为Score,小提琴图按照"Type"列("Low"和"High")进行填充。同时,添加盒图在小提琴图上,#并指定盒图的颜色为白色。这样可以同时展示数据的分布和中位数等统计信息。#输出图形pdf(file="vioplot.pdf", width=6, height=5)print(p1)dev.off()#使用pdf函数将绘制好的小提琴图保存为名为"vioplot.pdf"的PDF文件。#这个图形显示了不同肿瘤微环境得分组("Low"和"High")之间的差异,#并使用星号标记不同组之间的显著性差异(根据wilcox.test的p值)。#通过这个小提琴图,可以直观地展示肿瘤微环境得分与基因表达量之间的关联。
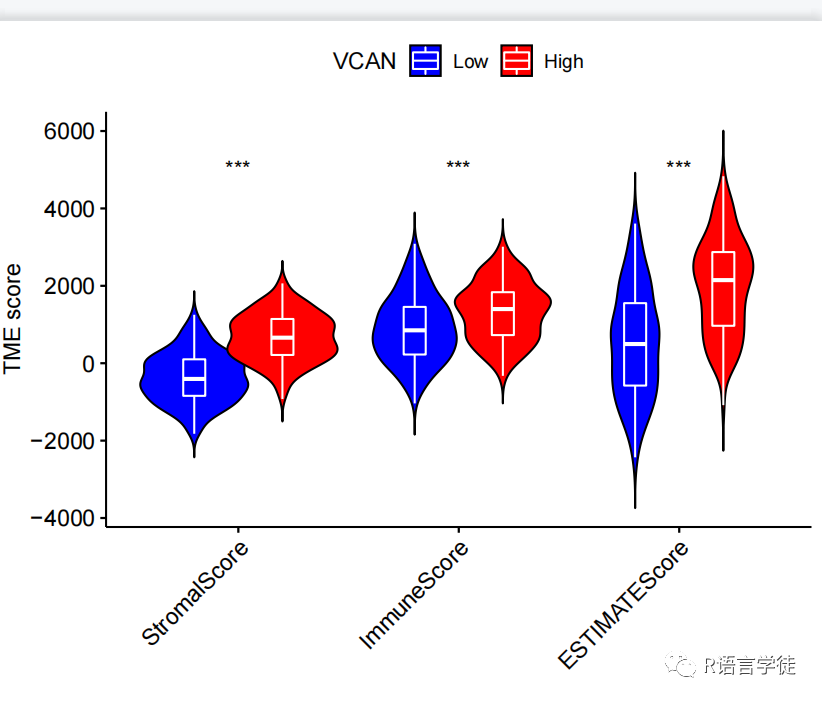
肿瘤微环境得分与基因表达量之间的关联。通过绘制小提琴图,可以直观地观察不同得分组之间基因表达数据的差异,进而了解肿瘤微环境得分在不同基因表达模式中的分布情况。这种关联分析可以为肿瘤研究提供重要的线索,帮助发现与肿瘤微环境相关的重要基因或通路,为深入研究肿瘤发生和发展提供有益的信息。
此外,小提琴图的可视化呈现使得结果更易于解释。分享下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:http://www.biocloudservice.com/home.html。
云生信平台链接:http://www.biocloudservice.com/home.html。
E
N
D

