比blast还优秀的序列比对工具?HMMER来了
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

小果今天给大家带来的分享绝对是生信热点分析内容,基于三种机器学习算法进行生存资料特征基因筛选,最终将三种算法获得的交叉基因作为我们的特征基因。
在日常的生信分析中,我们常见的序列比对工具基本都是blast进行比对。但是从研究基因功能的角度来讲,HMMER的准确性更高。而且HMMER在处理大规模序列数据时表现出色。它采用了优化的算法和数据结构,能够快速比对大量序列数据,节省时间和计算资源。看到这里,是不是很想知道如何使用HMMER了。别着急,小果带你来学习!
1. 什么是HMMER?
HMMER是一种常用的序列比对工具,它是基于隐马尔可夫模型(Hidden Markov Model,HMM)的算法。HMMER可以用于比对不同类型的生物序列,包括蛋白质序列和核酸序列。它可以根据不同的生物信息学问题进行定制,从而适应各种分析需求。
与此同时,HMMER可以利用隐马尔可夫模型进行比对,能够更好地处理复杂的序列间关系,提供较为准确的匹配结果。它能够发现较为相似的序列片段,捕获更多的生物信息。
当然,HMMER不仅可以进行序列比对,还能够进行序列分类、序列注释和特征预测等任务。它提供了丰富的功能和工具,帮助研究者深入分析和理解生物序列的特征和功能。
2. 如何使用HMMER?
首先我们可以在pfam数据库搜索p450蛋白质,p450蛋白是一类存在于生物体中、具有重要生物催化功能的酶。它们参与细胞内多种化学反应,包括代谢内源性物质和外源性药物、合成生理活性物质以及解毒等过程。在搜索之后,得到如下的结果,我们点击Accession为PF00067的蛋白。
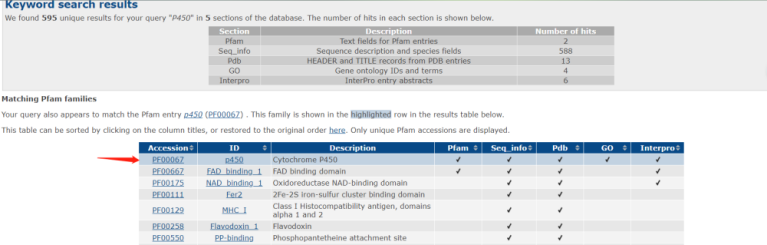
接着点击Alignments按钮,来到比对页面,接着我们选择stockholm格式,点击Generate按钮。此时,页面将会自动下载一个多序列比对文件PF00067_seed.txt。
(ps:私信小果“111”获取相关文件哦~)
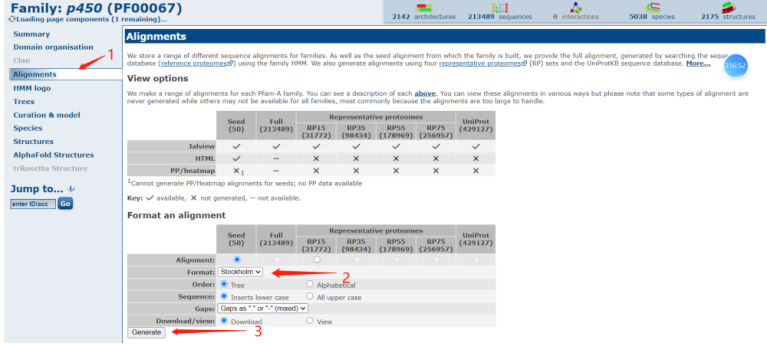
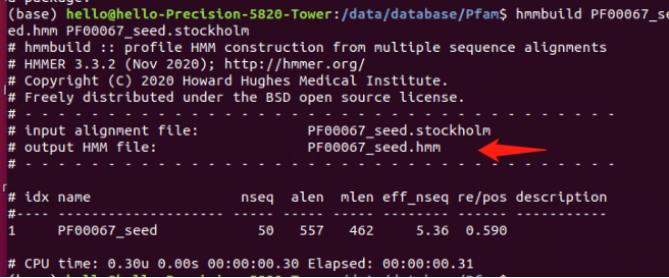
接着我们需要将把下载的文件扩展名.txt格式更改成.stockholm格式,并且放在hmmer程序的文件夹内;接着在终端中,使用HMMER的hmmbuild命令将域模型构建起来,该命令将域序列和相关信息作为输入,生成一个HMM文件。如图所示,我们采用了hmmbuild PF00067_seed.hmm PF00067_seed.stockholm指令,生成了一个hmm文件。
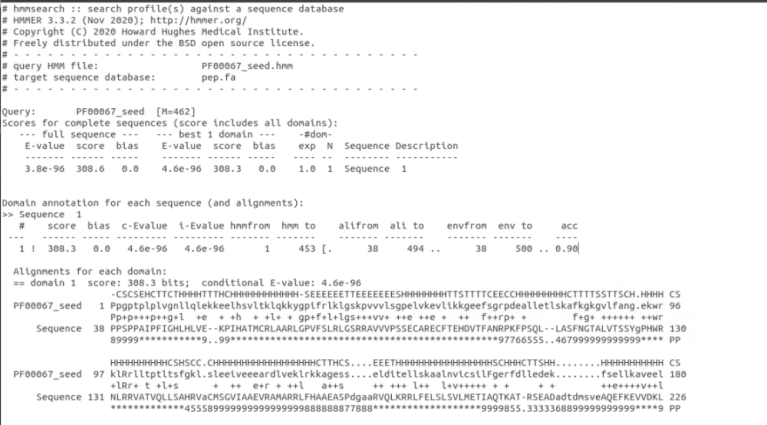
接下来我们将使用hmmsearch指令,输入hmmsearch PF00067_seed.hmm Icpep.fa > p450.out,该命令的意思是将目标序列文件 “pep.fa” 与保守域模型文件 “PF00067_seed.hmm” 进行比对,并将比对结果保存到名为 “p450.out” 的文件中。

该命令的输出结果将包含比对的相关信息,如匹配位置、得分等。可以打开 “p450.out” 文件之后,我们可以查看比对结果以及蛋白质序列是否包含与保守域模型相匹配的区域。
最后相信大家已经了解如何利用pfam数据库和HMMER进行比对啦!是不是能很切实地感受到HMMER非常高效便捷。
对了,如果大家还对其他生信的工具感兴趣的话,欢迎大家点击http://www.biocloudservice.com/home.html进行学习哦!
点击“阅读原文”立刻拥有
↓↓↓