DESeq2包做差异分析,简单易懂,有手就行,你还不学?
{ 点击蓝字,关注我们 }
转录组的主要目的之一便是寻找基因表达的差异解释基因表达水平的变化对生物功能的影响。例如,在癌变和其他复杂疾病发生和发展过程中,细胞内的基因表达模式会发生显著改变。在细菌和病毒侵染时,细胞内的基因表达模式也会发生显著变化,这些变化对机体的抗感染功能至关重要。在植物的正常生长,抗旱、抗逆、以及优良品系培育等过程中细胞的基因表达模式会发生显著变化。
那么,如何基于转录组测序获得的定量表达值,识别差异表达变化的基因或其它非编码RNA分子呢?
转录组测序完成后,一般我们会获得一个原始 read count表达矩阵,其中行是基因,列是样品。常用的差异分析工具包括limma,edgeR,DESeq2。小师妹就以R包DESeq2的差异表达基因分析方法为例做个简单演示,这是目前使用频率最高的鉴定差异分子的R包之一。
简单地说,DESeq2将对原始reads count进行建模,使用标准化因子(scale factor)来解释库深度的差异。然后DESeq2估计基因的离散度,并缩小这些估计值以生成更准确的离散度估计,从而对reads count进行建模。最后,DESeq2拟合负二项分布的模型,并使用Wald检验或似然比检验进行假设检验。
只需要知道怎么跑,准备数据,修改数据参数就ok了!
这里小师妹就不过多介绍这个包啦,主要是以出结果为目的。
所以直接上代码
一
包的准备
install.packages(‘BiocManager’) #需要首先安装 BiocManager,如果尚未安装请
install.packages('BiocManager')#需要首先安装 BiocManager,如果尚未安装请先执行该步BiocManager::install('DESeq2')library("DESeq2")#测试是否安装成功
二
准备数据
这里准备counts文件即可
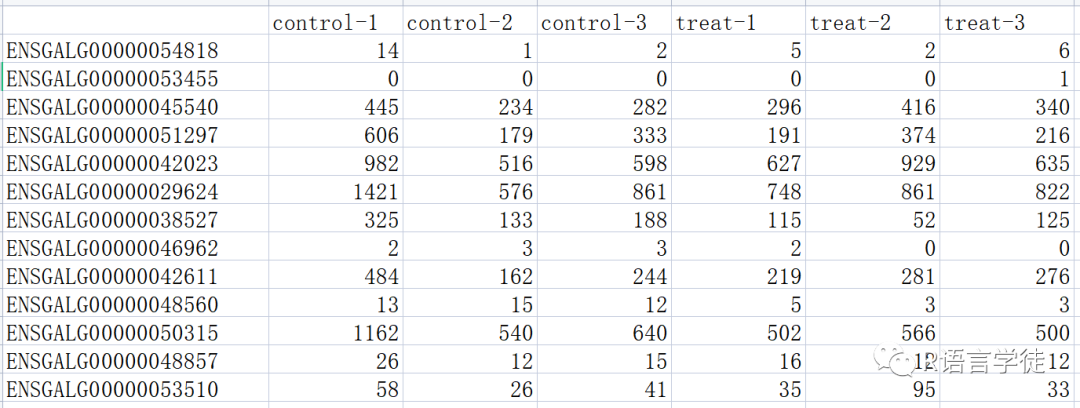
“mRNA_count.txt”,是6个测序样本的基因表达值矩阵,包括3个处理组(treat)和3个对照组(control)。第1列是基因名称,注意不能有重复值。
三
数据处理
然后将准备好的基因表达值矩阵读到R中,并预定义样本分组信息
result=read.table("mRNA_count.txt",header=T,seq=”t”,check.names=F) #读取基因表达矩阵rownames(result)=result[,1]result <- result [which(rowSums(result) > 0),]#数据预处理,去除在全部样本皆表达为0的基因group_list <- c(rep("control",times = 3),rep("treat",times = 3))#指定样本分组exprSet=result
四
进行差异分析
coldata <- data.frame(row.names = colnames(exprSet),group_list=group_list) #每一行对应一个样本,行名与countData的样本顺序一一对应,列为各种分组信息。dds<-DESeqDataSetFromMatrix(countData=exprSet,colData=coldata,design=~group_list) #构建DESeqDataSet对象dds<-DESeq(dds) #标准化res<-results(dds)#将结果用results()函数来获取,赋值给res变量class(res)#查看res数据类型res<-as.data.frame(res) #用data.frame转化res格式为表格形式res<-cbind(rownames(res),res)head(res)#查看构建好的表达矩阵res <- results(dds,contrast = c("group_list","treat","control"))#查看基因的差异倍数,和显著性p值。treat在前,control在后,意为treat相较于control中哪些基因上调/下调。res0rdered <- res[order(res$padj),]#对p值重新进行排序head(res0rdered)#查看res0rderedDEG=as.data.frame(res0rdered)DEG <- na.omit(DEG)# 去除缺失值NADEG<-cbind(rownames(DEG),DEG)head(DEG)colnames(res)<-c('gene_id',"baseMean" ,"log2FoldChange","lfcSE" ,"stat","pvalue","padj" )#res中,包含了基因id、标准化后的基因表达值(baseMean)、log2转化后的差异倍数(Fold Change)值、显著性p值以及校正后p值(默认FDR校正)等主要信息。write.table(res,"control-mRNA.xls",sep = 't',col.names = T,row.names = F,quote = FALSE,na='')#文件保存,得到完整表格
五
进一步筛选差异基因,使用Padj值和log2FC进行筛选
#获取padj(p值经过多重校验校正后的值)小于0.05,表达倍数取以2为对数后大于1或者小于-1的差异表达基因。resSig<-res[which(res$padj<0.05 & abs(res$log2FoldChange>1)),]resSig[which(resSig$log2FoldChange>0),'up_down']<-'up'#表达量显著上调的基因resSig[which(resSig$log2FoldChange<0),'up_down']<-'down'#表达量显著下调的基因head(resSig)write.table(resSig,"control-mRNA-0.05.xls",sep='t',col.names=T,row.names=F,quote = FALSE,na='')# 输出差异表达分析结果
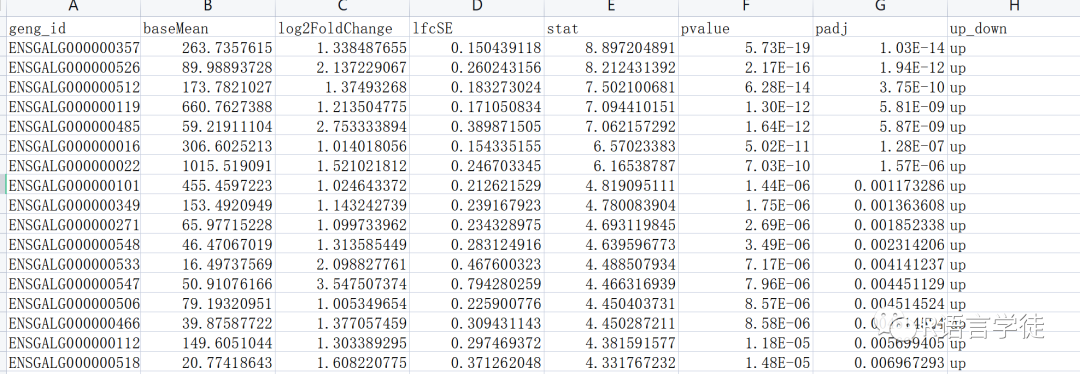
初学者最好先按照这个方法一步一步走,包括命名。每一步出结果都查看一下,如此便会顺利些呦!
好了小师妹今天的分享就到这啦,小伙伴们有遇到问题欢迎随时来找小师妹分享讨论呀!
欢迎使用:云生信 – 学生物信息学 (biocloudservice.com)
如果想用服务器可以联系微信:18502195490(快来联系我们使用吧!)
E
N
D
