转录组分析软件该选谁——质控篇
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

-
在学习转录组分析时,你是否会被铺天盖地的软件搞得眼花缭乱呢,学习完一个再学习另一个,最后发现两者功能竟然是一样的!甚至有些软件已经不再适用于转录组分析,可是依然出现在推荐的教程里。今天小果就带领大家擦亮眼睛,告别“老腊肉”,迎接“小鲜肉”!
在介绍软件之前,小果先带大家复习一下转录组分析的流程。转录组分析是一种根据不同条件下基因表达水平的差异来研究基因功能的方法。在分析之前要准备好转录组测序数据、参考基因组(有参)、基因组注释文件,在这里小果要提醒一句参考基因组注释文件一定要和参考基因组是相同版本,不然容易报错哦!
有了数据后就可以进行后续的质控、比对、表达分析、差异分析以及富集分析。下面小果带大家来挑选每个环节使用的软件。
数据质控
原始数据质量的好坏直接决定了是否能得到理想结果,在数据质控环节常用的软件有FastQC、MultiQC、Trimmomatic、Cutadapt、FASTX_Toolkit, FastQC可以对测序原始数据进行质量检验,生成质量检测报告,是最常用的质控软件。
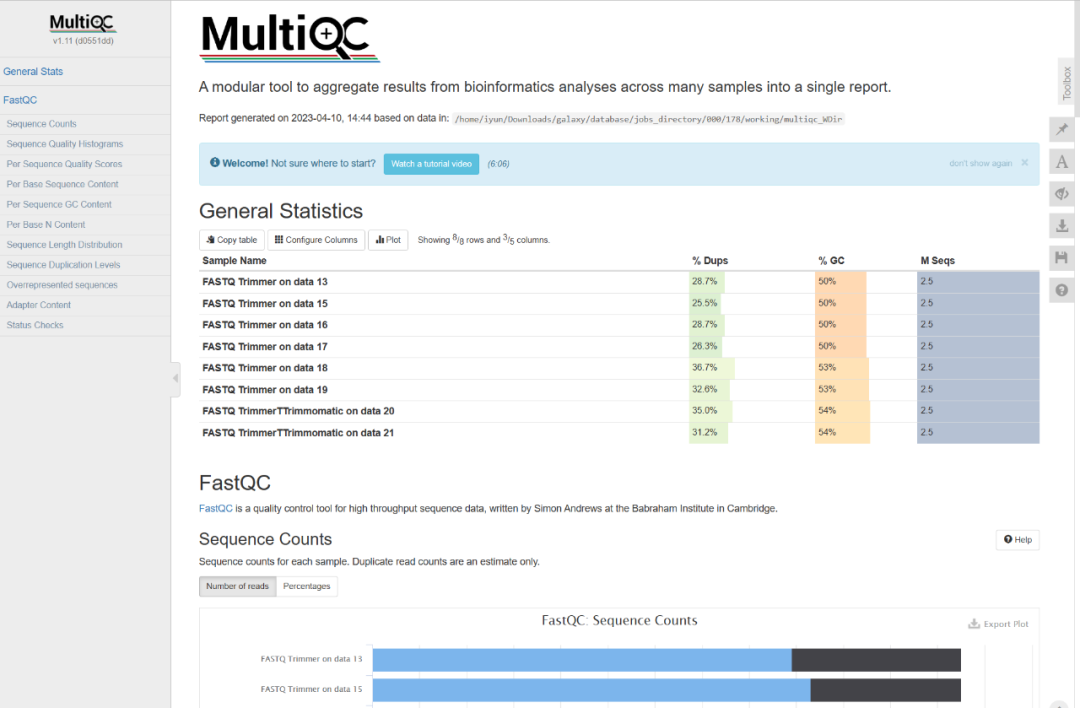
MultiQC工具可以将多个单独的FastQC结果整合为一个文件,方便统一查看同一批测序结果。
Trimmomatic、Cutadapt、FASTX_Toolkit三者均为对测序结果进行处理的软件,下面小果来给大家详细介绍一下这三位“神秘人”。
Trimmomatic
Trimmomatic适用于illumina二代测序数据的reads处理,主要对接头(adapter)序列和低质量序列进行过滤。Trimmomatic的参数并不多,从字面意思可以明白含义,自v0.32版本之后可自动识别碱基编码格式是phred33还是phred64。优点是操作简单、参数易懂。
FASTX-Toolkit
FASTX-Toolkit是用于短读FASTA / FASTQ文件预处理的命令行工具的集合。新一代测序数据通常包含多个短读序列。在将序列映射到基因组之前预处理FASTA / FASTQ文件有时能够提高效率从而得到更好的结果。但是要注意FASTX-Toolkit不支持压缩格式的输入文件;不允许序列中存在N碱基,这样的序列会自动去除;默认情况下认为FASTQ文件的碱基编码格式为phred64。总而言之FASTX-Toolkit参数众多,如果能够掌握可以提高工作效率,缺点就是容易报错,对小白不友好。
Cutadapt
Cutadapt是一个比较经典的能够对双端进行接头切除的软件,也可以删除primer、 polyA尾序列以及低质量序列。Cutadapt在去除接头方面非常专业,使用时要懂得一点测序原理,参数稍微有些复杂。
经过小果的讲解大家是否对转录组分析质控方面有了更多地了解呢,欢迎来和小果讨论哦!

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓