scRNA-seq数据分析求生指南之DoubletFinder:寻找双细胞的神秘之旅
 大家好,我叫小果,你的贴身学习助手,能够满足你的所有分析需求。今天果果要给你介绍一个非常实用的R包,叫做DoubletFinder。
大家好,我叫小果,你的贴身学习助手,能够满足你的所有分析需求。今天果果要给你介绍一个非常实用的R包,叫做DoubletFinder。
你可能已经知道,为了解决生物学的复杂问题,研究人员已经开始转向单细胞测序技术。这项技术能够帮助我们在单个细胞级别上理解基因表达的微妙差异,揭示生物体的更深层次的复杂性。然而,这项技术也有它的陷阱。其中之一就是会形成双细胞(doublets),即两个细胞在准备测序过程中不小心被混在一起,在分析中它们依旧会被当成同一个细胞的数据来处理。这会导致测序数据混乱,对数据的解读造成困难,所以你一定要认真学习如何使用DoubletFinder来消灭它们!
DoubletFinder是一个强大的工具,可以帮助你在单细胞转录组测序(scRNA-seq)数据中发现并剔除双细胞。那么,让我们一起踏上寻找单细胞数据中双细胞的神秘之旅吧!
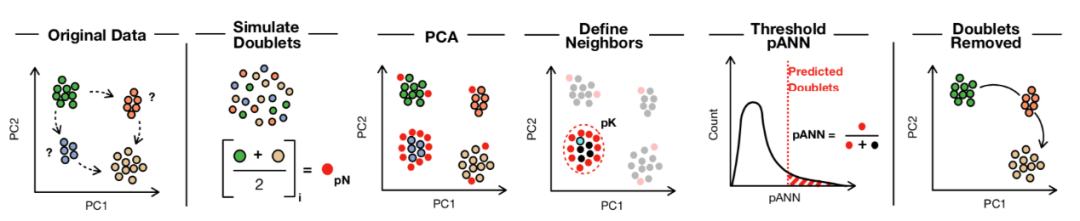 DoublebletFinder基础原理及步骤
DoublebletFinder基础原理及步骤
从现有的单细胞表达数据中随机将两个细胞的基因表达数据相加来,模拟可能出现的doublet的基因表达情况,生成人工模拟的双细胞。这些模拟出来的doublet会被当作正常的细胞进行后续的处理。
将人工模拟的双细胞和原始数据合并,进行预处理和常规的PCA降维。
计算每个细胞在主成分空间中的最近邻(nearest neighbors),并统计其中有多少是人工模拟的双细胞,得到每个细胞的人工最近邻比例(pANN, proportion of artificial nearest neighbors)。
根据预期的双细胞比例和数量,设置pANN的阈值,高于设定的值即鉴定为双细胞并进行移除。
听起来是不是so easy?那么就跟小果一起动手实战吧!
DoublebletFinder实战
首先,你需要安装DoubletFinder这个R包。可以用小果前面介绍的神器devtools从GitHub上下载并安装它:
devtools::install_github('chris-mcginnis-ucsf/DoubletFinder')安装好了之后,就可以加载这个包了:
library(DoubletFinder) 这里我们选择了一套包含4000多个细胞的人类肾脏样本作为示例数据:
这里我们选择了一套包含4000多个细胞的人类肾脏样本作为示例数据:
https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSM5225906&format=file&file=GSM5225906%5Fs200929N8%2Eexpression%5Fmatrix%2Etxt%2Egz# 导入数据, 是表达矩阵的压缩文件。data <- read.table(file="GSM5225906_s200929N8.expression_matrix.txt.gz")# 导入所需的库library(Seurat)#进行常规的数据预处理,创建一个Seurat对象(scRNA)。scRNA <- CreateSeuratObject(counts = data, min.cells = 3,min.features = 200, project = "kidney")scRNA <- SCTransform(scRNA)scRNA <- RunPCA(scRNA, verbose = F)scRNA <- RunUMAP(scRNA, dims = 1:15)scRNA <- FindNeighbors(scRNA, dims = 1:15) %>% FindClusters(resolution = 0.6)
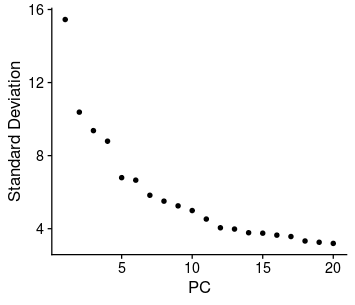
这里我们已经有了一个名为scRNA的Seurat对象,并且对它进行了标准化、主成分分析和UMAP降维等步骤。
#设置参数,寻找最优pK值
此时需要确定一些参数,比如主成分(PC)的个数、人工双细胞的比例(pN)、最近邻居的比例(pK)和预期的双细胞数量(nExp)。这些参数都会影响DoubletFinder的效果,所以我们要根据自己的数据特点来合理地选择它们。
#对scRNA这个单细胞对象进行pN-pK参数扫描,以生成人工双细胞并计算每个细胞的pANN值,这一步运行耗时最久。sweep.res.list <- paramSweep_v3(scRNA, PCs = 1:15, sct = T)#对参数扫描的结果进行汇总,计算每种pN和pK组合下的双细胞检测指标。这些指标可以用来评估不同参数下的双细胞检测效果,并选择最优的参数。参数GT表示是否提供了真实的双细胞标签,这里没有提供。sweep.stats <- summarizeSweep(sweep.res.list, GT = FALSE)#对汇总结果按照真实双胞胎比例(BCreal)进行升序排序,并显示排序后的数据框。这可以帮助我们找到双胞胎检测效果最好的参数组合sweep.stats[order(sweep.stats$BCreal),]#根据汇总结果找到最优的pK参数值。bcmvn <- find.pK(sweep.stats)#从上一步得到的数据框中提取出全局最优的pK值,也就是使得所有pN下BCmetric最大化的pK值。pK_bcmvn <- as.numeric(as.vector(bcmvn$pK[which.max(bcmvn$BCmetric)]))#根据单细胞数据的细胞数量来估计双细胞的比例。DoubletRate表示每5000个细胞中有多少个双细胞。这里假设每增加1000个细胞,双细胞比率就增加千分之8,这是一个经验值,可以根据实际情况调整。DoubletRate = ncol(scRNA)*8*1e-6#估计单细胞数据中同源双细胞的比例。同源双细胞是指由两个或多个相同类型的细胞融合而成的假阳性细胞,它们通常比异源双细胞更难以检测和去除。homotypic.prop表示同源双细胞的比例。函数modelHomotypic会根据给定的聚类标签来模拟生成一些同源双细胞,并计算它们在所有双细胞中所占的比例。homotypic.prop <- modelHomotypic(scRNA$seurat_clusters)#根据估计的双细胞比例和同源双细胞比例来计算期望的异源双细胞数量。异源双细胞是指由两个或多个不同类型的细胞融合而成的假阳性细胞。nExp_poi表示期望的总双细胞数量,它等于估计的双细胞比例DoubletRate乘以单细胞对象scRNA中的总细胞数量nrow(scRNA@meta.data),并四舍五入取整。nExp_poi.adj表示期望的异源双细胞数量。nExp_poi <- round(DoubletRate*nrow(scRNA@meta.data))# 使用同源双细胞比例对推算出期望的异源双细胞数量nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))##使用DoubletFinder算法来检测和去除单细胞数据中的异源双细胞。doubletFinder_v3函数会对单细胞对象scRNA进行双细胞检测和鉴定。这个函数有以下参数:PCs:表示要使用的主成分数量,这里是1到15。pN:表示每个细胞被合并成人工异源双细胞的概率,这里是0.25。pK:表示每个细胞的最近邻居数量,这里使用之前计算出来的最优值。nExp:表示期望的总双细胞数量,这里使用之前计算出来的值。
 reuse.pANN:表示是否重用之前计算出来的pANN值,pANN是每个细胞的最近邻居中人工异源双细胞的比例。
reuse.pANN:表示是否重用之前计算出来的pANN值,pANN是每个细胞的最近邻居中人工异源双细胞的比例。
sct:表示是否使用了SCTransform方法进行标准化。
scRNA <- doubletFinder_v3(scRNA, PCs = 1:15, pN = 0.25, pK = pK_bcmvn, nExp = nExp_poi.adj, reuse.pANN = F, sct = T)## 运行结果储存在scRNA@meta.data中
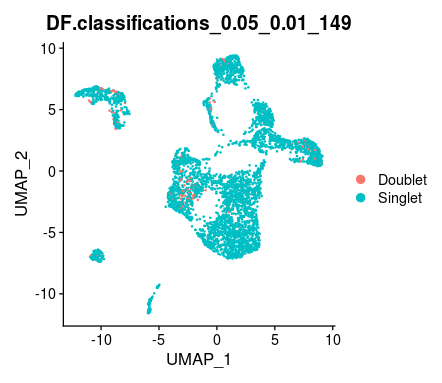
#可视化如图
#将双细胞剔除后生成新的对象scRNA.singlet,便于我们后续分析
scRNA.singlet <- subset(scRNA, subset = DF.classifications_0.25_0.01_129== "Singlet")至此,你已经完成了寻找双细胞的神秘之旅。希望你在这次旅程中收获满满,通过DoubletFinder,你已经学会了如何在单细胞RNA测序数据中找出并去除那些可能扰乱数据分析的双细胞,让我们得到更加纯净和可靠的数据。
今天小果的陪伴就到这里啦,下次我们还会继续带来更多有趣的科学知识,敬请期待!


往期推荐