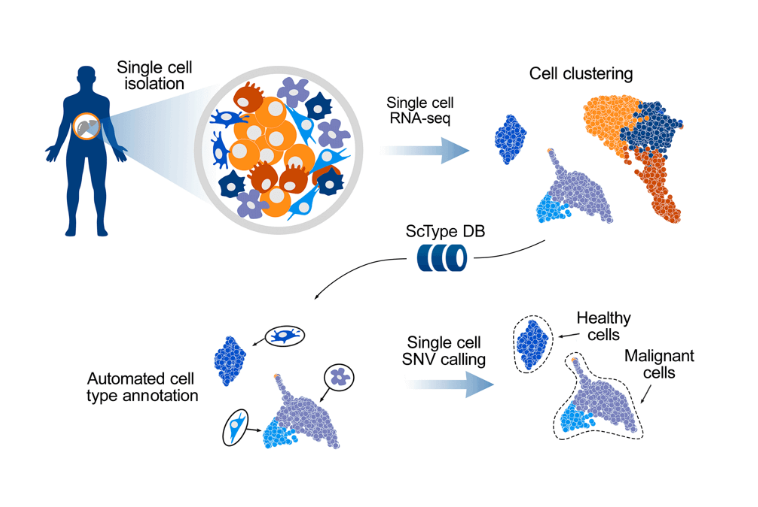
小果带你玩转单细胞注释工具-ScType

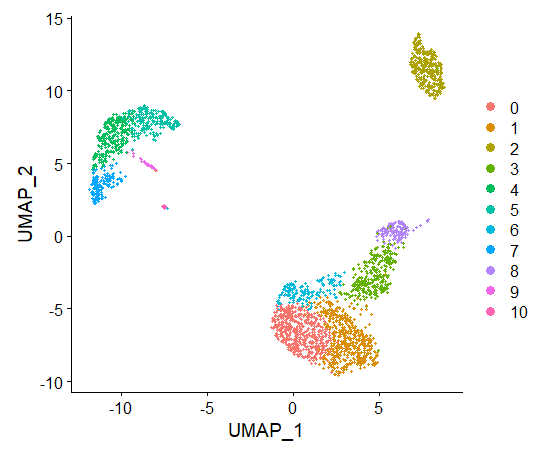
#设置工作环境,这里要改成自己的工作环境哦!!!!setwd("D:\work")# 加载R包lapply(c("dplyr","Seurat","HGNChelper"), library, character.only = T)#数据下载:https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz# 加载PBMC数据集pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")# Initialize the Seurat object with the raw (non-normalized data).pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)# 数据标准化pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")# pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5) # make some filtering based on QC metrics visualizations, see Seurat tutorial: https://satijalab.org/seurat/articles/pbmc3k_tutorial.htmlpbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)# 进行PCA处理pbmc <- ScaleData(pbmc, features = rownames(pbmc))pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))# 检查 PC 组件的数量(根据 Elbow 图,我们选择了 10 个 PC 进行下游分析)ElbowPlot(pbmc)# 聚类和可视化pbmc <- FindNeighbors(pbmc, dims = 1:10)pbmc <- FindClusters(pbmc, resolution = 0.8)pbmc <- RunUMAP(pbmc, dims = 1:10)DimPlot(pbmc, reduction = "umap")
##细胞类型注释# 使用 ScType 自动分配单元格类型。首先加载两个包#载入基因组构建函数source("https://raw.githubusercontent.com/IanevskiAleksandr/sc-type/master/R/gene_sets_prepare.R")# 加载细胞类型注释source("https://raw.githubusercontent.com/IanevskiAleksandr/sc-type/master/R/sctype_score_.R")
#DB filedb_ = "https://raw.githubusercontent.com/IanevskiAleksandr/sc-type/master/ScTypeDB_full.xlsx";tissue = "Immune system" # e.g. Immune system,Pancreas,Liver,Eye,Kidney,Brain,Lung,Adrenal,Heart,Intestine,Muscle,Placenta,Spleen,Stomach,Thymus# prepare gene setsgs_list = gene_sets_prepare(db_, tissue)# prepare gene setsgs_list = gene_sets_prepare(db_, tissue)# get cell-type by cell matrixes.max = sctype_score(scRNAseqData = pbmc[["RNA"]]@scale.data, scaled = TRUE,gs = gs_list$gs_positive, gs2 = gs_list$gs_negative)
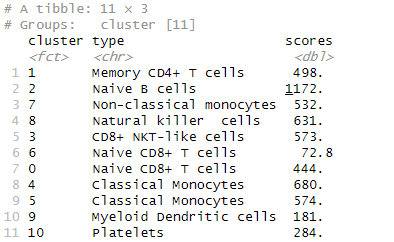
# 按分群合并cL_resutls = do.call("rbind", lapply(unique(pbmc@meta.data$seurat_clusters), function(cl){es.max.cl = sort(rowSums(es.max[ ,rownames(pbmc@meta.data[pbmc@meta.data$seurat_clusters==cl, ])]), decreasing = !0)head(data.frame(cluster = cl, type = names(es.max.cl), scores = es.max.cl, ncells = sum(pbmc@meta.data$seurat_clusters==cl)), 10)}))sctype_scores = cL_resutls %>% group_by(cluster) %>% top_n(n = 1, wt = scores)# 将低置信度(ScType 分数低)群设为 "unknown"sctype_scores$type[as.numeric(as.character(sctype_scores$scores)) < sctype_scores$ncells/4] = "Unknown"print(sctype_scores[,1:3])
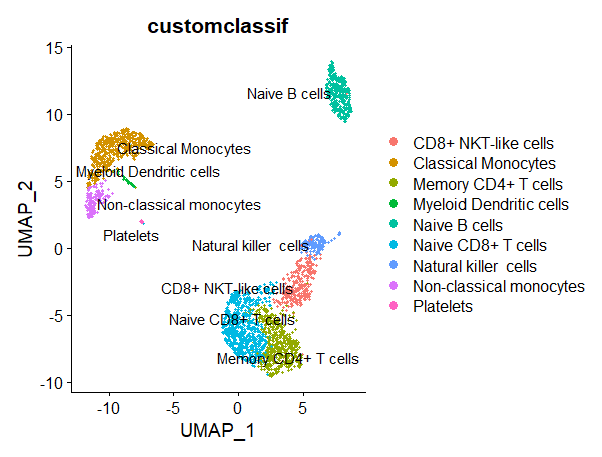
##可视化#将识别的细胞类型叠加在 UMAP 图上pbmc@meta.data$customclassif = ""for(j in unique(sctype_scores$cluster)){cl_type = sctype_scores[sctype_scores$cluster==j,];pbmc@meta.data$customclassif[pbmc@meta.data$seurat_clusters == j] = as.character(cl_type$type[1])}DimPlot(pbmc, reduction = "umap", label = TRUE, repel = TRUE, group.by = 'customclassif')
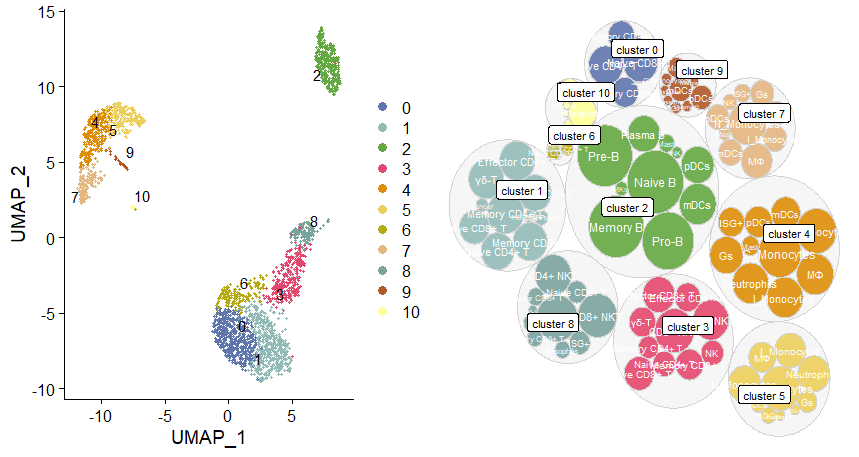
# 加载R包lapply(c("ggraph","igraph","tidyverse", "data.tree"), library, character.only = T)# 绘图cL_resutls=cL_resutls[order(cL_resutls$cluster),]; edges = cL_resutls; edges$type = paste0(edges$type,"_",edges$cluster); edges$cluster = paste0("cluster ", edges$cluster); edges = edges[,c("cluster", "type")]; colnames(edges) = c("from", "to"); rownames(edges) <- NULLnodes_lvl1 = sctype_scores[,c("cluster", "ncells")]; nodes_lvl1$cluster = paste0("cluster ", nodes_lvl1$cluster); nodes_lvl1$Colour = "#f1f1ef"; nodes_lvl1$ord = 1; nodes_lvl1$realname = nodes_lvl1$cluster; nodes_lvl1 = as.data.frame(nodes_lvl1); nodes_lvl2 = c();ccolss= c("#5f75ae","#92bbb8","#64a841","#e5486e","#de8e06","#eccf5a","#b5aa0f","#e4b680","#7ba39d","#b15928","#ffff99", "#6a3d9a","#cab2d6","#ff7f00","#fdbf6f","#e31a1c","#fb9a99","#33a02c","#b2df8a","#1f78b4","#a6cee3")for (i in 1:length(unique(cL_resutls$cluster))){dt_tmp = cL_resutls[cL_resutls$cluster == unique(cL_resutls$cluster)[i], ]; nodes_lvl2 = rbind(nodes_lvl2, data.frame(cluster = paste0(dt_tmp$type,"_",dt_tmp$cluster), ncells = dt_tmp$scores, Colour = ccolss[i], ord = 2, realname = dt_tmp$type))}nodes = rbind(nodes_lvl1, nodes_lvl2); nodes$ncells[nodes$ncells<1] = 1;files_db = openxlsx::read.xlsx(db_)[,c("cellName","shortName")]; files_db = unique(files_db); nodes = merge(nodes, files_db, all.x = T, all.y = F, by.x = "realname", by.y = "cellName", sort = F)nodes$shortName[is.na(nodes$shortName)] = nodes$realname[is.na(nodes$shortName)]; nodes = nodes[,c("cluster", "ncells", "Colour", "ord", "shortName", "realname")]mygraph <- graph_from_data_frame(edges, vertices=nodes)gggr<- ggraph(mygraph, layout = 'circlepack', weight=I(ncells)) +geom_node_circle(aes(filter=ord==1,fill=I("#F5F5F5"), colour=I("#D3D3D3")), alpha=0.9) + geom_node_circle(aes(filter=ord==2,fill=I(Colour), colour=I("#D3D3D3")), alpha=0.9) +theme_void() + geom_node_text(aes(filter=ord==2, label=shortName, colour=I("#ffffff"), fill="white", repel = !1, parse = T, size = I(log(ncells,25)*1.5)))+ geom_node_label(aes(filter=ord==1, label=shortName, colour=I("#000000"), size = I(3), fill="white", parse = T), repel = !0, segment.linetype="dotted")scater::multiplot(DimPlot(pbmc, reduction = "umap", label = TRUE, repel = TRUE, cols = ccolss), gggr, cols = 2)
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |