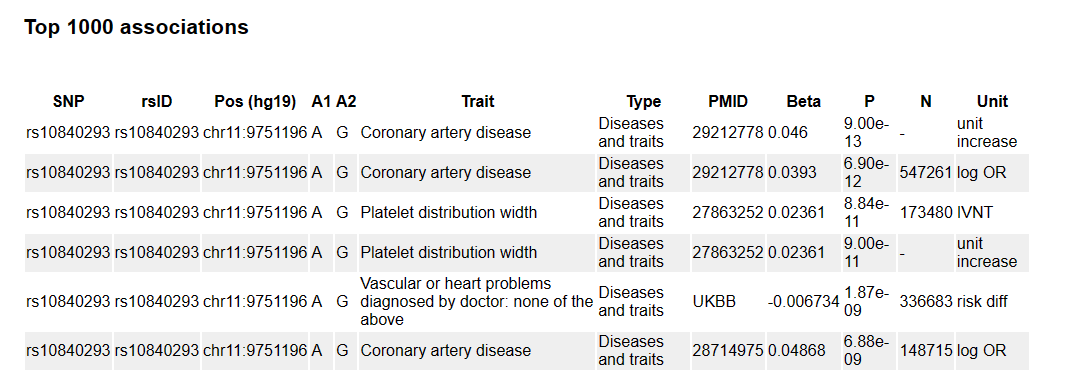
孟德尔随机化——如何选择工具变量 小图 生信果 2024-01-07 19:00:34 转自公众号:生信图http://mp.weixin.qq.com/s?__biz=MzkxODUxNDgzNA==&mid=2247486090&idx=1&sn=5300cc70cee096f765b70fb417e3ae16 一、表型GWAS结果中具有显著关联的SNP 标准阈值小于5e-8显著位点(比如SNP位点数大于20个),可以直接进行后续的筛选 如果阈值小于5e-8显著位点只有个位数,甚至为0,那么可以尝试1e-7、1e-6、1e-5作为阈值筛选条件 注:1e-5阈值需要后续敏感性分析和异质性性检验结果增加解释依据。 二、去除连锁不平衡(LD) 设置r2=0.001和kb=10000 表示去掉在10000kb范围内与最显著SNP的r2大于0.001的SNP 设置成r2=0.3和kb=1000 表示的就是去掉在1000kb范围内与最显著SNP的r2大于0.3的SNP 参数设置r2=0.01, kb=5000即可,如在上一步基础上SNP过滤较多,也可调整参数(r2=0.1, kb=5000) 三、筛选MAF >1% MAF肯定是<0.5的,因为如果>0.5,那就不是第二多。在GWAS的summary文件里经常没有MAF这一列,但是有EAF这一列,他们之间的关系就是:如果EAF>0.5,那么MAF=1-EAF;如果EAF<0.5,那么MAF=EAF。 四、去除弱工具变量 F检验值>10(一般F统计量大于10是比较好的,当然能大于100是更好的) 计算方法 1.方法一 其中N表示GWAS分析中的样本量;k 表示工具变量的个数;R^ {2} 表示工具变量解释暴露因素的程度。 2.方法二 β2/SE2 注:如果方法一F值皆小于10,可尝试方法二计算 五、剔除混杂SNPs影响(上述步骤筛选后,SNP数目较多可做此步,反之不建议做) 利用PhenoScanner 网站链接:http://www.phenoscanner.medschl.cam.ac.uk/ 首先打开网站 根据筛选到的SNP进行检索排除 在此处输入要查找SNP的名字 下滑界面可浏览检索到相应性状结果 例如暴露因素是失眠 结局因素是心脏病 检索结果发现此SNP与心血管疾病有关,那么这个SNP可能是通过心血管疾病影响的心脏病,因此需要查阅文献排除是否此SNP与心血管疾病有关,如有建议剔除此SNP。 如果SNP数据量较多可以上传文本文件批量查询 注:SNP数目不要超过100个 最后提醒一下,SNP数目多的时候统计效力足,但是异质性和多效性可能会比较大,如果去除部分SNP后,可以消除异质性和多效性,但是会导致统计效力低,使结果变成阴性。因此,大家需要好好斟酌一下自己的筛选阈值条件。上述选取原则具有一定普适性,希望大家记住,感兴趣的可以深入理解一下为什么遵循这些原则。 想要更好的学习和交流,快来加入小图的微信公众号(生信图)和云生信生物信息学平台( http://www.biocloudservice.com/home.html),在这里你可以向小图提问、帮你制定相应分析操作。点击这里加入吧! 往期推荐 1.搭建生信分析流水线,如工厂一样24小时运转Snakemake——进阶命令 2.比blast还优秀的序列比对工具?HMMER来了 3.对单细胞分析毫无头绪?让popsicleR领你入门 4.小果带你绘制ROC曲线评估生存预测能力 5.软件包安装、打怪快又好,1024G存储的生信服务器;还有比这更省钱的嘛!!!
 一、表型GWAS结果中具有显著关联的SNP
一、表型GWAS结果中具有显著关联的SNP
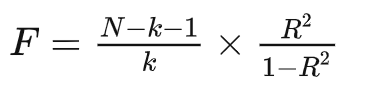

 一、表型GWAS结果中具有显著关联的SNP
一、表型GWAS结果中具有显著关联的SNP