单细胞表型预测黑科技!3分钟带你解锁ScPP完全体!!

 在进行教程之前,我们首先需要了解什么是单细胞表型预测,单细胞表型预测(Single Cells’Phenotype Prediction)是一种利用单细胞数据来推断细胞的功能或状态的方法,可以帮助研究人员发现细胞的异质性、转化关系,并且有助于揭示细胞表型与基因型、蛋白质以及环境因素之间的关联,还可以应用于药物响应、疾病诊断、组织发育和干细胞分化等。可以说是单细胞分析中非常具有开发潜能的一个方向呢。
在进行教程之前,我们首先需要了解什么是单细胞表型预测,单细胞表型预测(Single Cells’Phenotype Prediction)是一种利用单细胞数据来推断细胞的功能或状态的方法,可以帮助研究人员发现细胞的异质性、转化关系,并且有助于揭示细胞表型与基因型、蛋白质以及环境因素之间的关联,还可以应用于药物响应、疾病诊断、组织发育和干细胞分化等。可以说是单细胞分析中非常具有开发潜能的一个方向呢。
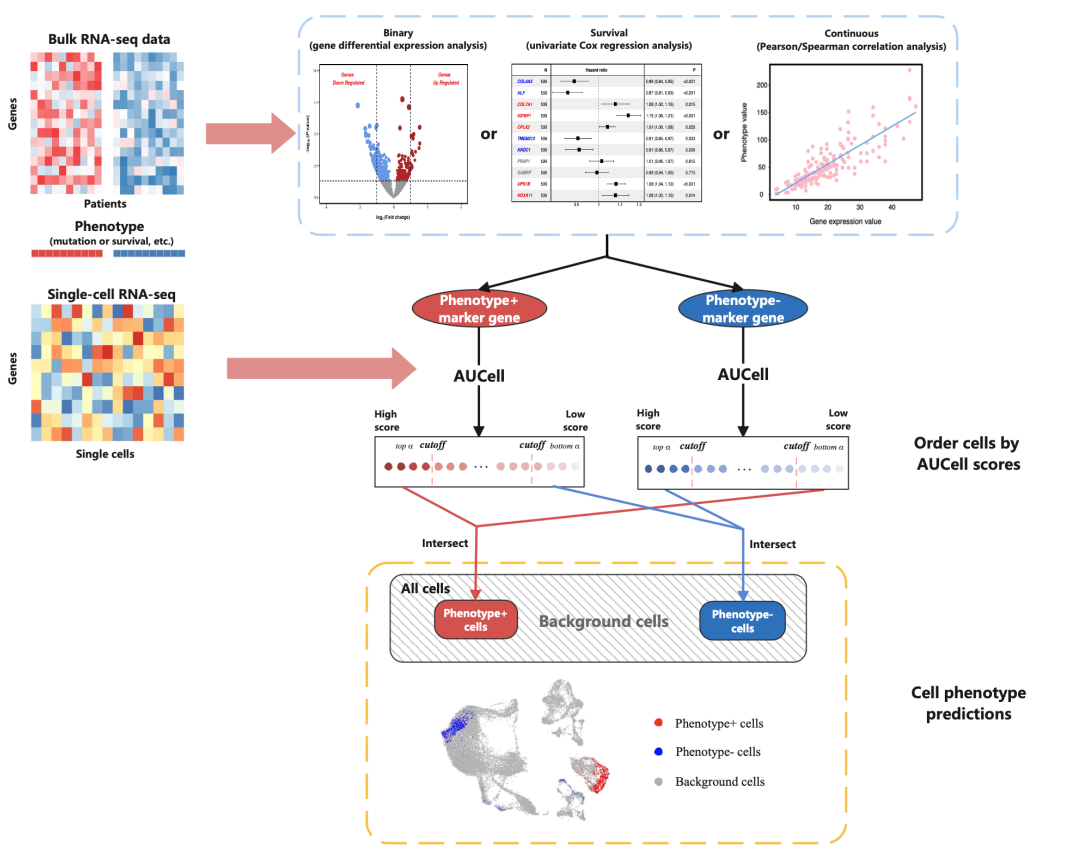 |
#导入ScPP中的功能函数##为单细胞表达数据进行预处理。输入数据是一个计数矩阵,其中细胞是列名,基因是行名sc_Preprocess <- function(counts, project = "sc_preprocess",normalization.method = "LogNormalize", scale.factor = 10000,selection.method = "vst", resolution = 0.1,dims_Neighbors = 1:20, dims_TSNE = 1:20, dims_UMAP = 1:20){library(Seurat)sc_dat <- CreateSeuratObject(counts = counts, project = project)sc_dat <- NormalizeData(object = sc_dat, normalization.method = normalization.method, scale.factor = scale.factor)sc_dat <- FindVariableFeatures(object = sc_dat, selection.method = selection.method)sc_dat <- ScaleData(object = sc_dat, features = rownames(sc_dat))sc_dat <- RunPCA(object = sc_dat, features = VariableFeatures(sc_dat))sc_dat <- FindNeighbors(object = sc_dat, dims = dims_Neighbors)sc_dat <- FindClusters(object = sc_dat, resolution = resolution)sc_dat <- RunTSNE(object = sc_dat, dims = dims_TSNE)sc_dat <- RunUMAP(object = sc_dat, dims = dims_UMAP)return(sc_dat)}#运用Binary进行差异表达分析##生成二元变量与表型+或表型-组相关的标记基因以及特征,一共包含4个参数:bulk_data、features、ref_group和Log2FC_cutoffmarker_Binary <- function(bulk_data, features, ref_group, Log2FC_cutoff = 0.585){library(dplyr)if (missing(ref_group))stop("'ref_group' is missing or incorrect.")if (missing(bulk_data) || !class(bulk_data) %in% c("matrix", "data.frame"))stop("'bulk_data' is missing or incorrect.")if (missing(features) || !class(features) %in% c("matrix", "data.frame"))stop("'features' is missing or incorrect.")ref = features$Sample[features$Feature == ref_group]tes = features$Sample[features$Feature != ref_group]ref_pos = which(colnames(bulk_data) %in% ref)tes_pos = which(colnames(bulk_data) %in% tes)pvalues <- apply(bulk_data, 1, function(x) {t.test(as.numeric(x)[tes_pos], as.numeric(x)[ref_pos])$p.value})log2FCs <- rowMeans(bulk_data[, tes_pos]) - rowMeans(bulk_data[, ref_pos])res <- data.frame(pvalue = pvalues, log2FC = log2FCs)res <- res[order(res$pvalue), ]res$fdr <- p.adjust(res$pvalue, method = "fdr")geneList <- list(gene_pos = res %>% filter(pvalue < 0.05, log2FC > Log2FC_cutoff ) %>% rownames(.),gene_neg = res %>% filter(pvalue < 0.05, log2FC < -Log2FC_cutoff ) %>% rownames(.))if(length(geneList[[1]]) > 0 & length(geneList[[2]] > 0)){return(geneList)}else if (length(geneList[[1]]) == 0){warning("There is no genes positively correlated with the given feature in this bulk dataset.");geneList = list(gene_pos = geneList[[2]]);return(geneList)}else if (length(geneList[[2]]) == 0){warning("There is no genes negatively correlated with the given feature in this bulk dataset.");geneList = list(gene_neg = geneList[[1]]);return(geneList)}}#进行连续变量相关性分析##生成连续变量以及和表型相关的标记基因或特征,包含4个参数:bulk_data、features、method和estimate_cutoffmarker_Continuous <- function(bulk_data, features, method = "spearman", estimate_cutoff = 0.2){library(dplyr)if (missing(bulk_data) || !class(bulk_data) %in% c("matrix", "data.frame"))stop("'bulk_data' is missing or incorrect.")if (missing(features) || !class(features) %in% c("integer", "numeric"))stop("'features' is missing or incorrect.")CorrelationTest = apply(bulk_data,1,function(x){pvalue = cor.test(as.numeric(x),log2(as.numeric(features)+1), method = method)$p.valueestimate = cor.test(as.numeric(x),log2(as.numeric(features)+1), method = method)$estimateres = cbind(pvalue,estimate)return(res)})resc = t(CorrelationTest)colnames(resc) = c("pvalue","estimate")resc = as.data.frame(resc[order(as.numeric(resc[,1]),decreasing = FALSE),])resc$fdr <- p.adjust(resc$pvalue,method = "fdr")geneList <- list(gene_pos = resc %>% filter(fdr < 0.05, estimate > estimate_cutoff) %>% rownames(.),gene_neg = resc %>% filter(fdr < 0.05, estimate < -estimate_cutoff) %>% rownames(.))if(length(geneList[[1]]) > 0 & length(geneList[[2]] > 0)){return(geneList)}else if (length(geneList[[1]]) == 0){warning("There is no genes positively correlated with the given feature in this bulk dataset.");geneList = list(gene_pos = geneList[[2]]);return(geneList)}else if (length(geneList[[2]]) == 0){warning("There is no genes negatively correlated with the given feature in this bulk dataset.");geneList = list(gene_neg = geneList[[1]]);return(geneList)}}##生存数据分析,生成与患者预后相关的标记基因或特征,包含2个参数:bulk_data和survival_datamarker_Survival <- function(bulk_data,survival_data){library(survival)library(dplyr)if (missing(bulk_data) || !class(bulk_data) %in% c("matrix", "data.frame"))stop("'bulk_data' is missing or incorrect.")if (missing(survival_data) || !class(survival_data) %in% c("matrix", "data.frame"))stop("'survival_data' is missing or incorrect.")SurvivalData <- data.frame(cbind(survival_data,t(bulk_data)))colnames(SurvivalData) = make.names(colnames(SurvivalData))var <- make.names(rownames(bulk_data))Model_Formula <- sapply(var, function(x) as.formula(paste("Surv(time, status) ~", x)))Model_all <- lapply(Model_Formula, function(x) coxph(x, data = SurvivalData))res <- lapply(seq_along(Model_all), function(i) {coef_summary <- summary(Model_all[[i]])$coefficientsdata.frame(variable = var[i],pvalue = coef_summary[,5],coef = coef_summary[,2])}) %>% bind_rows()res <- res[order(res$pvalue), ]res$fdr <- p.adjust(res$pvalue, method = "fdr")geneList <- list(gene_pos = res %>% filter(fdr < 0.05, coef > 1) %>% pull(variable), #correalted with worse survivalgene_neg = res %>% filter(fdr < 0.05, coef < 1) %>% pull(variable) #correlated with better survival)if(length(geneList[[1]]) > 0 & length(geneList[[2]] > 0)){return(geneList)}else if (length(geneList[[1]]) == 0){warning("There is no genes negatively correlated with patients' prognosis in this bulk dataset.");geneList = list(gene_pos = geneList[[2]]);return(geneList)}else if (length(geneList[[2]]) == 0){warning("There is no genes positively correlated with patients' prognosis in this bulk dataset.");geneList = list(gene_neg = geneList[[1]]);return(geneList)}}##进行单细胞表型预测,包含3个参数:sc_dataset, geneList和probs。ScPP = function(sc_dataset, geneList, probs = 0.2){if(length(geneList) != 2){stop("This gene list do not have enough information correlated with interested feature.")}if (missing(sc_dataset) || class(sc_dataset) != "Seurat")stop("'sc_dataset' is missing or not a seurat object.")library(AUCell)cellrankings = AUCell_buildRankings(sc_dataset@assays$RNA@data,plotStats = FALSE)cellAUC = AUCell_calcAUC(geneList,cellrankings)metadata = as.data.frame(sc_dataset@meta.data)metadata$AUCup <- as.numeric(getAUC(cellAUC)["gene_pos", ])metadata$AUCdown <- as.numeric(getAUC(cellAUC)["gene_neg", ])downcells1 = rownames(metadata)[which(metadata$AUCup <= quantile(metadata$AUCup,probs = probs))]upcells1 = rownames(metadata)[which(metadata$AUCup >= quantile(metadata$AUCup,probs = (1-probs)))]downcells2 = rownames(metadata)[which(metadata$AUCdown >= quantile(metadata$AUCdown,probs = (1-probs)))]upcells2 = rownames(metadata)[which(metadata$AUCdown <= quantile(metadata$AUCdown,probs = probs))]ScPP_neg = intersect(downcells1,downcells2)ScPP_pos = intersect(upcells1,upcells2)metadata$ScPP <- ifelse(rownames(metadata) %in% ScPP_pos, "Phenotype+", "Background")metadata$ScPP <- ifelse(rownames(metadata) %in% ScPP_neg, "Phenotype-", metadata$ScPP)return(metadata)}#导入绘图所需的包library(ggplot2)#安装Scpp包以及相关的R包if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("AUCell")if (!requireNamespace("devtools", quietly = TRUE))install.packages("devtools")devtools::install_github("WangX-Lab/ScPP")
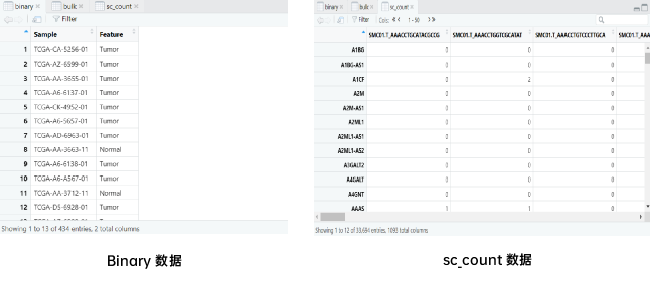
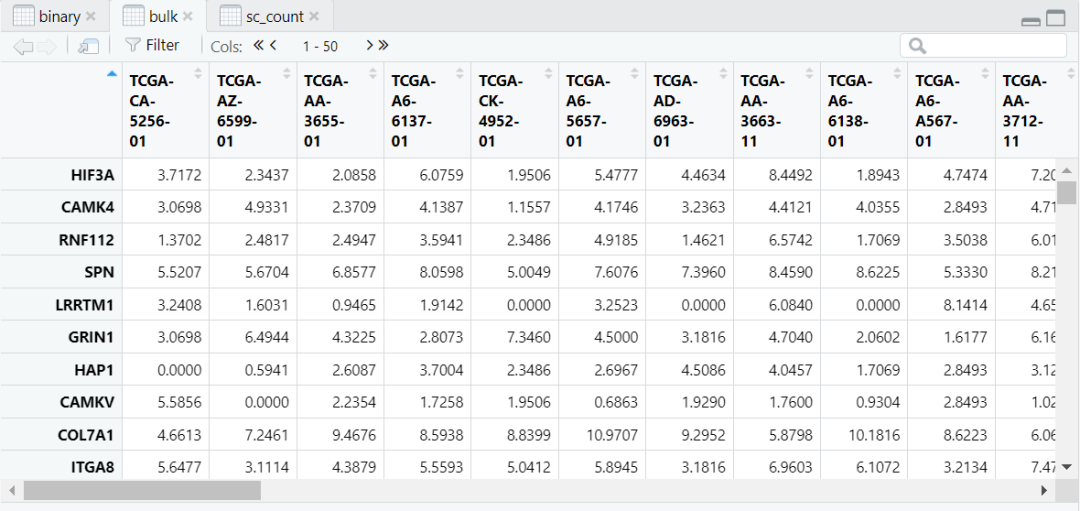
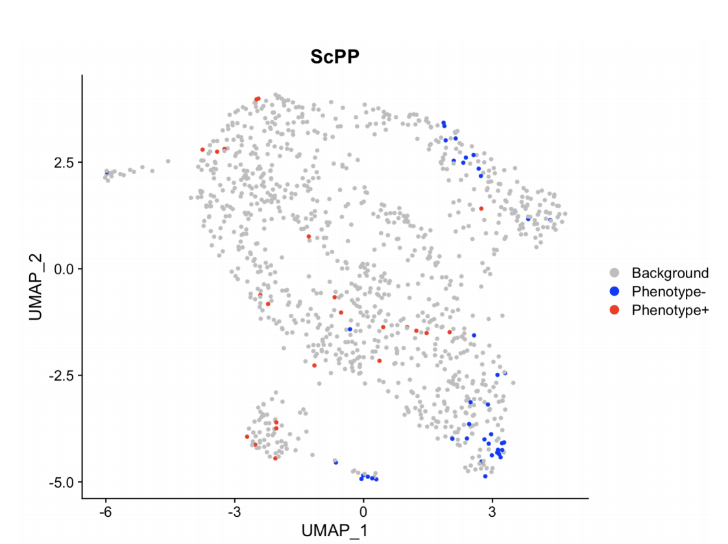
# 准备数据load("/data/binary.RData")# 数据总览bulk[1:6,1:6] # bulk datahead(binary) # phenotype data of bulksc_count[1:6,1:6] # scRNA-seq data# 通过 ScPP 选择信息细胞sc = sc_Preprocess(sc_count)geneList = marker_Binary(bulk, binary, ref_group = "Normal")metadata = ScPP(sc, geneList)write.csv(metadata, "/results/binary_meta.csv")head(metadata)sc$ScPP = metadata$ScPPIdents(sc) = "ScPP"#对ScPP识别的细胞进行可视化DimPlot(sc, group = "ScPP", cols = c("grey","blue","red"))ggsave("Fig_binary.pdf",path = "/results/",width = 10, height = 10, units = "cm")
 |
 |
 |
 |
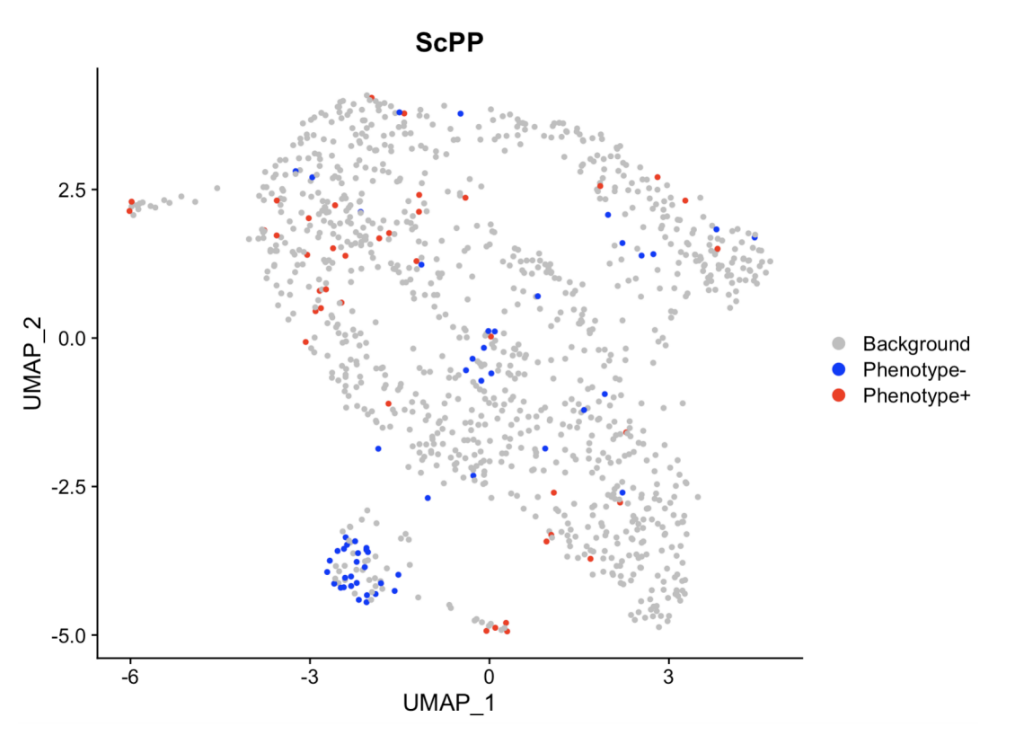
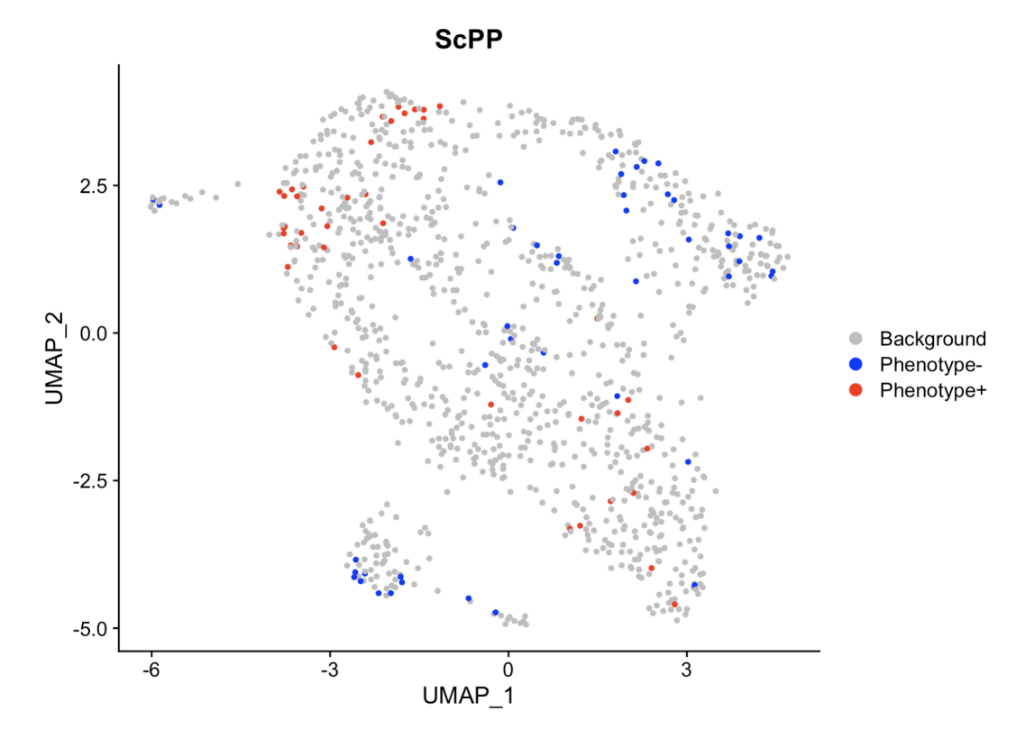
# 导入数据(所需数据小果放在上文的链接了哦~)# library(ScPP)load("/data/continuous.RData")# 数据总览bulk[1:6,1:6] # bulk datahead(continuous) # phenotype data of bulksc_count[1:6,1:6] # scRNA-seq data# 通过 ScPP 选择信息细胞sc = sc_Preprocess(sc_count)geneList = marker_Continuous(bulk, continuous$TMB_non_silent)metadata = ScPP(sc, geneList)write.csv(metadata, "/results/continuous_meta.csv")sc$ScPP = metadata$ScPPIdents(sc) = "ScPP"#对ScPP识别的细胞进行可视化DimPlot(sc, group = "ScPP", cols = c("grey","blue","red"))ggsave("Fig_continuous.pdf",path = "/results/",width = 10, height = 10, units = "cm")
 |
 |
#导入数据load("/data/survival.RData")#通过 ScPP 选择信息细胞sc = sc_Preprocess(sc_count)geneList = marker_Survival(bulk, survival)str(geneList)metadata = ScPP(sc, geneList)write.csv(metadata, "/results/survival_meta.csv")sc$ScPP = metadata$ScPPIdents(sc) = "ScPP"#可视化DimPlot(sc, group = "ScPP", cols = c("grey","blue","red"))ggsave("Fig_survival.pdf",path = "/results/",width = 10, height = 10, units = "cm")
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |