SnpEff:一个神奇的基因组变异注释工具
点击蓝字 关注我们
在生物信息学中,分析基因组变异是一个重要的任务。变异可以是单核苷酸多态性(SNP)、插入缺失(indel)或结构变异(SV)。变异的类型和影响不同,可能导致基因表达、蛋白质功能或表型的改变。因此,对变异进行注释和解释是理解其生物学意义的关键步骤。那如何实现对不同变异类型的注释呢?
目前,有许多工具可以用来注释变异,但其中一个比较流行和强大的是snpEFF。snpEFF是一个开源的Java程序,可以对VCF格式的变异文件进行快速和准确的注释。snpEFF可以预测变异对转录本和蛋白质的影响,例如错义、无义、剪切位点、启动子、终止子等。snpEFF还可以提供其他有用的信息,例如基因名、转录本ID、外显子编号、密码子改变、氨基酸改变等;snpEFF支持包括人类、动物、植物、微生物等超过38,000个物种的基因组数据库,也可以自定义数据库或使用公共数据库。支持多种变异格式,包括VCF、GFF、BED等;支持多种输出格式,包括文本、HTML、VCF等;支持多种统计分析,包括变异频率、变异类型分布、变异功能分布等。
snpEFF的使用非常简单,只需要几个命令行参数就可以运行。snpEFF的使用包括以下几步,首先需要下载snpEFF和相应的基因组数据库或者自定义数据库。然后使用snpEff命令对VCF文件进行注释,指定数据库名称和输出文件名。最后,可以使用snpEff的伴随工具SnpSift对注释结果进行过滤、排序或统计。snpEFF还可以与其他工具集成,例如GATK、bcftools或VEP。
1. snpEFF的下载与安装
方法一,下载压缩包安装。下载网址:https://sourceforge.net/projects/snpeff/
下载解压后,会在snpEff文件夹中生产两个jar包snpEff.jar和SnpSift.jar。运行命令java -jar snpEff.jar 生成帮助文件则安装成功。
方法二,使用conda安装snpEFF。
conda是可以方便地安装和更新各种软件。要使用conda安装snpEFF,首先需要安装conda,可以参考官方网站的教程:https://docs.conda.io/en/latest/miniconda.html 。安装好conda后,打开终端,输入以下命令:
conda install -c bioconda snpeff
这个命令会从bioconda频道下载并安装snpEFF及其依赖的软件包。安装完成后,可以使用以下命令检查snpEFF的版本:
snpeff -version
如果显示SnpEff版本信息,说明snpEFF已经成功安装。
2. 下载或构建数据库
方法一:使用snpEFF自带的数据库
snpEFF自带了一些常见物种的数据库,如人类、小鼠、大肠杆菌等,这些数据库可以直接下载使用。下载的命令如下:
java -jar snpEff.jar download -v hg19
其中hg19是人类基因组的版本号,可以根据需要替换为其他物种和版本。下载完成后,可以在snpEff/data目录下看到相应的文件夹,里面包含了基因组序列和注释信息。
方法二:使用GFF3或GTF文件建库
如果snpEFF没有提供所需物种的数据库,或者需要使用自己的注释信息,可以使用GFF3或GTF文件来建库。这些文件可以从各种数据库或网站下载,也可以自己生成。使用GFF3或GTF文件建库的命令如下:
java -jar snpEff.jar build -c snpEff.config -gff3 -v my_genome其中my_genome是自定义的数据库名称,可以随意指定。-c指定配置文件snpEff.config,-v指定冗长模式。-gff3表示使用GFF3格式的文件,如果是GTF格式,则改为-gtf。在执行命令之前,需要先在snpEff/data目录下创建一个以my_genome为名的文件夹,并将基因组序列和GFF3或GTF文件放入其中。基因组序列必须命名为my_genome.fa,GFF3或GTF文件必须命名为genes.gff或genes.gtf。并在snpEff.config配置文件最后一行加入:my_genome.genome : my_genome。
3. snpEFF的使用方法
snpEFF的基本用法是:
java -jar snpEff.jar [options] genome_version input_file > output_file其中:- genome_version:指定要使用的参考基因组和注释数据库的版本,如GRCh38.99、hg19等。可以使用`java -jar snpEff.jar databases`命令查看可用的数据库列表。- input_file:指定要注释的变异文件,可以是VCF、GFF、BED等格式,也可以是标准输入(stdin)。- output_file:指定输出文件,可以是标准输出(stdout),也可以是一个文件名。输出文件默认是VCF格式,也可以通过参数指定其他格式。snpEFF的常见参数和选项snpEFF有很多参数和选项,可以根据需要进行调整。以下是一些常见的参数和选项:- -v:显示详细信息,包括程序版本、数据库版本、运行时间等。- -noLog:不输出日志信息。- -noStats:不输出统计信息。- -no-downstream:不注释下游区域(默认为5kb)的变异。- -no-upstream:不注释上游区域(默认为5kb)的变异。- -no-intergenic:不注释间基因区域的变异。- -no-intron:不注释内含子区域的变异。- -no-utr:不注释UTR区域的变异。- -formatEff:输出格式为EFF格式,而不是VCF格式。EFF格式是一种简化的格式,只包含变异的影响和严重程度等信息。- -csvStats:输出统计信息为CSV格式,而不是HTML格式。CSV格式方便用其他软件进行分析和绘图。
示例命令:java -Xmx4G -jar snpEff.jar eff -csvStats variants.SnpEff.csv -s variants.SnpEff.html -c snpEff.config -v -ud 500 my_genome my_snp.vcf > my.SnpEff.vcf
-
-Xmx2G指定Java虚拟机的最大堆大小为4GB。
-
-csvStats variants.SnpEff.csv 指定输出CSV格式的统计信息。
-
-s variants.SnpEff.html 指定输出HTML格式的摘要报告。
-
-ud 500指定上游/下游区间长度为500个碱基。
-
my_genome 指定要使用的基因组。
-
my_snp.vcf 指定输入VCF文件。
结束后共输出4个文件:variants.SnpEff.csv、variants.SnpEff.html、my.SnpEff.vcf、my.SnpEff.genes.txt。4. snpEFF注释结果解读
HTML格式的摘要报告是一个网页文件,可以用浏览器打开。它包含了以下几个部分:
-
Summary:这部分显示了一些基本的信息,如基因组名称、注释日期、注释命令、警告信息、错误信息、输入文件行数、变异位点数(过滤前后)、非变异位点数、具有ID的变异位点数、非双等位基因组SNP位点数、Number of effects、参考基因组总长度、参考基因组有效长度、变异率等。

-
Variant types:这部分显示了不同类型的变异的数量和百分比,包括SNP(单核苷酸多态性)、MNP(多核苷酸多态性)、INS(插入变异)、DEL(缺失变异)、MIXED(混合变异)、INV (倒位变异)、DUP(重复变异)、BED(易位变异)、INTERVAL(间隔变异)等。

-
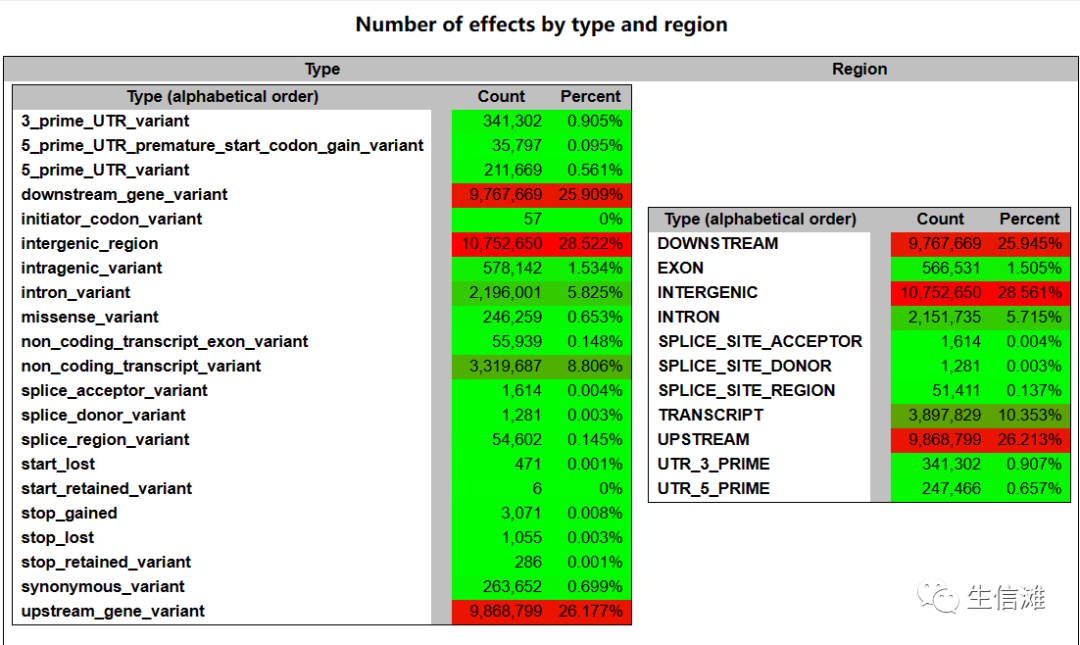
Effects by type:这部分显示了不同类型的效应的数量和百分比,包括3’端主要UTR变异、5’端主要UTR提前启动子获得变异、5’端主要UTR变异、下游基因变异、起始密码子编码变异、基因间隔区、内含子变异、剪接受体变异、剪接供体变异、剪接区域变异、起始缺失、起始保留变异、终止获得、终止缺失、终止保留变异、同义变异、上游基因变异等。
-
Effects by region:这部分显示了不同区域的效应的数量和百分比,包括下游、外显子、间隔区、内含子、剪接位点受体、剪接位点供体、剪接位点区域、上游、3’UTR区、5’UTR区等。

-
Effects by functional class:这部分显示了不同功能类别的效应的数量和百分比,包括无效果(NONE)、修饰性(MODIFIER)、低影响(LOW)、中等影响(MODERATE)、高影响(HIGH)等。

-
Transitions / Transversions:这部分显示了转换/颠换的比例和数量,以及不同类型的转换/颠换的数量和百分比。转换是指嘌呤与嘌呤或嘧啶与嘧啶之间的替代,颠换是指嘌呤与嘧啶之间的替代。

-
Allele frequency histogram:这部分显示了等位基因频率的直方图,横坐标为等位基因频率,纵坐标为数量。
-
Allele count histogram:这部分显示了等位基因数量的直方图,横坐标为等位基因数量,纵坐标为数量。
-
Genotype summary:这部分显示了样本基因型的汇总信息,包括杂合基因型(HETEROZYGOUS)、纯合基因型(HOMOZYGOUS)、缺失基因型(MISSING)等。
-
Codon change histogram:这部分显示了密码子改变的直方图,横坐标为密码子改变类型,纵坐标为数量。密码子改变是指单核苷酸突变导致密码子发生改变。
-
Amino acid change histogram:这部分显示了氨基酸改变的直方图,横坐标为氨基酸改变类型,纵坐标为数量。氨基酸改变是指单核苷酸突变导致密码子发生改变,进而导致氨基酸发生改变(错义突变)。
-
Chromosome summary:这部分显示了染色体的汇总信息,包括染色体名称、参考基因组长度、有效长度、变异位点数、变异率等。
snpEFF是一个非常强大而又方便的注释工具,我强烈推荐大家尝试一下。如果你想了解更多关于snpEFF的信息,你可以访问它的官网:http://snpeff.sourceforge.net/。如果你有任何关于snpEFF的问题或建议,你可以在评论区留言,我会尽快回复你。谢谢大家的阅读,下次再见!
大海哥今天的讲解就到这里了~下期我将会带来对单细胞测序分析结果的解读~我们不见不散
(最后推荐一下大海哥新开发的零代码云生信分析工具平台包含超多零代码小工具,上传数据一键出图,感兴趣的小伙伴欢迎来参观哟。
网址:
http://www.biocloudservice.com/home.html)

生信滩公众号


点击“阅读原文”进入网址