干货批量处理泛癌相关临床相关分析
点击蓝字,关注我们
临床相关分期是指将患者的疾病分为不同的阶段或分期,以便评估疾病的严重程度、预后以及指导治疗决策。临床相关分期在医学领域中具有重要意义,主要包括以下几个方面:
预后评估:临床相关分期可以帮助医生评估患者疾病的预后,即疾病的发展和治疗后的预期结果。通过将患者分为不同的阶段,医生可以更好地了解疾病的进展速度、转移倾向以及可能的生存期。治疗指导:临床相关分期可以为医生提供指导,帮助确定最合适的治疗方案。不同分期的患者可能需要不同的治疗策略,包括手术、放疗、化疗或靶向治疗等。研究和临床试验:通过对患者进行分期,研究人员可以更好地比较不同分期患者的疗效和生存结果。这有助于评估新的治疗方法、药物或治疗策略的有效性,并为进一步的研究提供基础。
肿瘤临床阶段分析是对患者肿瘤发展阶段与其生存情况之间关系的研究。临床分期是将肿瘤分为不同阶段,以便评估患者的疾病进展和预测生存率。以下是进行肿瘤临床阶段分析的一般步骤:
数据准备:收集包含患者临床信息和生存数据的数据集。临床信息可能包括患者的性别、年龄、肿瘤类型、肿瘤大小、淋巴结转移情况等。
数据清洗:对数据进行清洗和预处理,包括处理缺失值、异常值和重复数据等。
临床分期:根据肿瘤类型和相关指南,将患者按照临床分期系统进行分组。常用的临床分期系统包括TNM分期、AJCC分期等。
结果解释:根据分析结果,解释不同临床分期组别之间的生存差异,评估临床分期在预测患者生存率和治疗策略制定中的意义。
#if (!requireNamespace("BiocManager", quietly = TRUE)# install.packages("BiocManager")#BiocManager::install("limma")#install.packages("ggpubr")library(limma)library(ggpubr)#首先,加载了一系列所需的R包files=dir("./TG_Exp/")
#使用dir("./TG_Exp/")获取./TG_Exp/目录下的文件列表,这些文件包含了每种癌症类型的数据。cli=read.table("./clinical_stage.txt",sep="t",header=T,check.names=F,row.names=1)#使用read.table读取名为clinical_stage.txt的文件,该文件包含了临床阶段的信息,并将第一列的列名保存到变量clinical中。clinical=colnames(cli)[1]for ( i in 1:length(files) ){dirname=files[i]#获取文件名保存到变量dirname中。rt=read.table(paste0("./TG_Exp/",dirname),header=T,sep='t',check.names=F,row.names=1)#使用read.table读取文件内容,并保存到变量rt中。rt=na.omit(rt)#使用na.omit函数删除数据中的缺失值。gene=colnames(rt)[1]#获取基因表达数据的列名保存到变量gene中。#FOR CANCERoutTab=data.frame()tmp<- strsplit(dirname,split=".",fixed=TRUE)dirnam<-unlist(lapply(tmp,head,1))tissue=c("ACC", "BLCA", "BRCA", "CESC", "CHOL", "COAD", "DLBC", "ESCA", "GBM", "HNSC", "KICH", "KIRC", "KIRP", "LGG", "LIHC", "LUAD", "LUSC", "MESO", "OV", "PAAD", "PCPG", "PRAD", "READ", "SARC", "SKCM", "STAD", "TGCT", "THCA", "THYM", "UCEC", "UCS", "UVM")#根据癌症类型进行循环分析。for ( j in 1:length(tissue) ){cancer=tissue[j]rt1=rt[(rt$tissue %in% cancer),]#根据癌症类型选择对应的数据,保存到变量rt1中。#INSERCT#将基因表达数据与临床信息数据进行交集操作。data=cbind(rt1,gene=rt1[,"tpm"])data=as.matrix(data[,c(gene,"tpm")])if(nchar(row.names(data)[1])!=nchar(row.names(cli)[1])){row.names(data)=gsub(".$","",row.names(data))}data=avereps(data)sameSample=intersect(row.names(data),row.names(cli))sameData=data[sameSample,]sameClinical=cli[sameSample,]cliExpData=cbind(as.data.frame(sameClinical),sameData)#将交集后的数据保存到变量cliExpData中if(nrow(cliExpData)==0){next}#如果cliExpData为空,则跳过当前循环,继续下一个癌症类型的分析。#COMPgroup=levels(factor(cliExpData$sameClinical))#创建分组信息,保存到变量group中comp=combn(group,2)#进行组间差异比较的多重比较。my_comparisons=list()for(j in 1:ncol(comp)){my_comparisons[[j]]<-comp[,j]}#boxplot#使用ggboxplot函数绘制盒须图,x轴为临床阶段信息,y轴为基因表达#数据,颜色区分不同临床阶段。boxplot=ggboxplot(cliExpData, x="sameClinical", y="tpm", color="sameClinical",xlab=clinical,ylab=paste(dirnam),legend.title=clinical,title=paste0("Cancer: ",cancer),add = "jitter")+stat_compare_means(comparisons = my_comparisons)#使用stat_compare_means函数进行组间差异比较,并将比较结果添加到盒#须图中。pdf(file=paste0("./stage/",clinical,"_",cancer,"_",dirnam,".pdf"),width=5.5,height=5)#使用pdf函数将盒须图保存为PDF文件。print(boxplot)dev.off()}}
通过以上代码,可以对基因表达数据进行盒须图绘制,并进行不同临床阶段之间的组间差异比较分析,结果以PDF文件的形式保存。有问题可以联系小师妹。
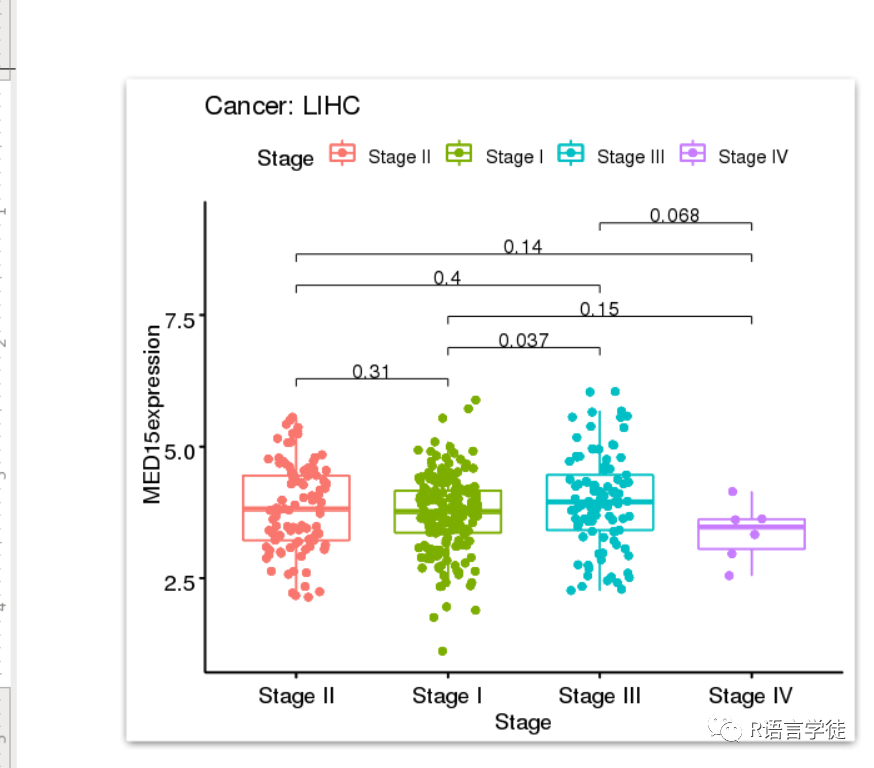
来给大家展示展示结果图吧,使用firefox命令。
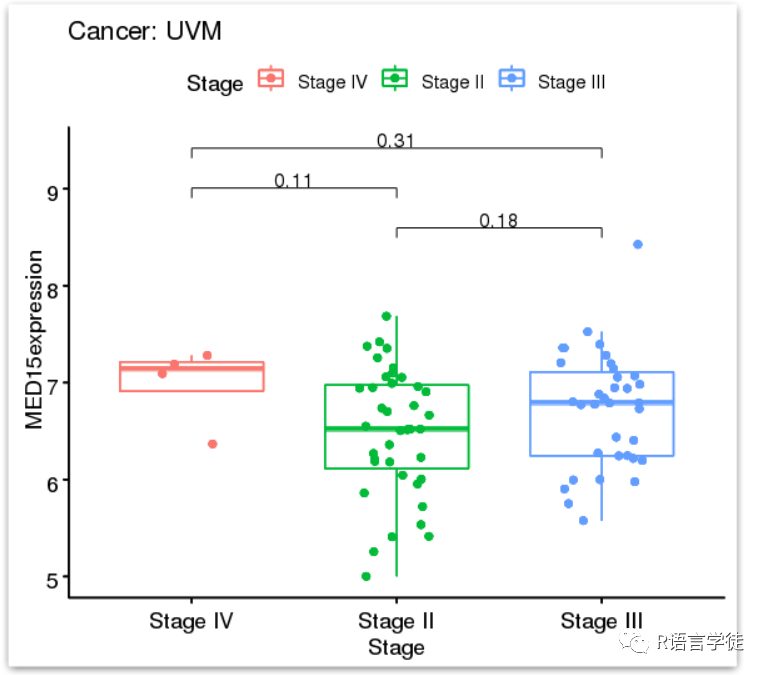
下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:
http://www.biocloudservice.com/home.html。
云生信平台链接:
http://www.biocloudservice.com/home.html。

END

